Bioinformatic Investigation of Micro RNA-802 Target Genes, Protein Networks, and Its Potential Prognostic Value in Breast Cancer
-
Eini, Maryam
-
Department of Biotechnology, Faculty of Allied Medicine, Iran University of Medical Sciences, Tehran, Iran
-
Parsi , Sepideh
-
Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School Worcester, MA, USA
-
Department of Biotechnology, Faculty of Allied Medicine, Iran University of Medical Sciences, Tehran, Iran
-
Kiani, Jafar
-
Department of Molecular Medicine, Faculty of Advanced Technologies in Medicine, Iran University of Medical Sciences, Tehran, Iran
-
Azarnezhad, Asaad
-
Liver and Digestive Research Center, Research Institute for Health Development, Kurdistan University of Medical Sciences, Sanandaj, Iran
-
 Hosseini, Arshad
Department of Biotechnology, Faculty of Allied Medicine, Iran University of Medical Sciences, Tehran, Iran, dr.arshdhoss96@gmail.com
Hosseini, Arshad
Department of Biotechnology, Faculty of Allied Medicine, Iran University of Medical Sciences, Tehran, Iran, dr.arshdhoss96@gmail.com
-
Department of Biotechnology, Faculty of Allied Medicine, Iran University of Medical Sciences, Tehran, Iran
Abstract: Background: An increasing number of studies have suggested that unveiling the molecular network of miRNAs may provide novel therapeutic targets or biomarkers. In this study, we investigated the probable molecular functions that are related to microRNA-802 (miR-802) and evaluated its prognostic value in breast cancer utilizing bioinformatics tools.
Methods: PPI network, pathway enrichment and transcription factor analysis were applied to obtain hub genes among overlapping genes of four miRNA target prediction databases. Prognosis value assessments and expression analysis of hub genes using bioinformatics tools, as well as their literature validation were performed.
Results: Our results showed a significant correlation of the miR-802 overexpression with poor patient survival rate (BC, p=2.7e-5). We determined 247 target genes significant for GO and KEGG terms. Analysis of TFs by TRUST showed that RUNX3, FOXO3, and E2F1 are possible TFs that regulate the miR-802 expression and target genes network. According to our analysis; 21 genes might have an important function in miR-802 molecular processes and regulatory networks. The result shows that among these 21 genes, 8 genes (CASC3, ITGA4, AGO3, TARDBP, MED13L, SF1, SNRPE and CRNKL1) are positively correlated with patient survival. Therefore these genes could be considered and experimentally evaluated as a prognostic biomarker for breast cancer.
Conclusion: The comprehensive bioinformatics study on miR-802 target genes provided insight into miR-802 mediated pathways and processes. Furthermore, representing candidate target genes by prognostic values indicates the potential clinical application of miR-802 in breast cancer.
Introduction :
By estimating 276,480 new cases in 2020 in the United States, breast cancer will be the most expected diagnosed malignancy in women that accounts for 30% of all new cancer occurrences in women. In addition, breast cancer will be the second cause of cancer deaths in women by estimating 42,690 deaths 1. Despite all progression in diagnostic and therapeutic approaches for breast cancer, it is estimated by WHO that the total worldwide cases will increase from 2,069,792 to 2, 778,850 cases between 2018 and 2040, that shows the necessity of more investigation on the molecular mechanisms of the disease as well as detecting more efficient biomarkers 2.
Recently molecular approaches have been mentioned for breast cancer classifications and understanding its underlying tumorigeneses mechanisms 3. Since now, alteration in signaling pathways, dysregulation of cells proliferation or apoptosis, mutation of oncogenes, different tumor metabolism, epithelial-to-mesenchymal transition, and Breast Cancer Stem Cells (BCSCs) development are among the most outlined molecular mechanisms 4-7.
Also many treatments, survival or follow-up investigations are fulfilled based on the molecular pattern of the patients 8,9. In this regard, prognostic markers are so important due to their use in the estimation of disease recurrence and treatment response. Predictions based on prognostic markers are used to determine the proper treatment strategy that has a direct effect on patient's life prolongation as well as recurrence-free or overall survival 10. A systematic review has demonstrated that the expression of PI3K, TIMP-1, CEACAM6, and aromatase are some reliable prognostic biomarkers for breast cancer 11. However, the poor outcome of the pa-tients, especially in the advanced stage, reflects the need for more investigation for prognostic markers which are more clinically applicable 12.
Since the discovery of 22 nucleotides noncoding microRNAs about two decades ago, the role of these molecules has been highlighted in different biological processes such as development, differentiation, apoptosis, cell proliferation, and so on 13-15. The microRNAs biological effect is exerted by post-translation regulation of about 30% of human genes, half of which are considered tumor-associated 16. Due to their fundamental regulatory roles, dysregulation of microRNAs profile may result in malignancies including oncogenic transformation 17. Regarding breast cancer, many studies have been conducted to identify diagnostic, prognostic, stage discriminating, or metastasis predictive biomarkers using aberrant expression profiles of microRNAs as well 18-21.
Recently it has been reported that an emerging microRNA, miR-802, has shown important regulatory roles in many malignancies including breast cancer. Previous investigations have revealed dis-regulation of miR-802 in the liver, gastric and cervical cancer 22-24. It has been reported that miR-802 inhibits epithelial-mesenchymal transition in human prostate cancer by targeting flotillin-2 25. Moreover, miR-802 targeted ZNF521 gene and suppressed the malignant progression of hepatocellular carcinoma 26. A study in breast cancer has demonstrated that miR-802 inhibits proliferation through the suppression of FoxM1 27. Regarding the prognostic value of miR-802, it is reported that patients with hepatocellular carcinoma and prostate cancer that have lower expression of miR-802 showed better survival 28. Despite the researches for determining miR-802 roles, there are not enough reports about the roles of miR-802 in the molecular basis of diseases and its clinical potential in malignancies as well as breast cancer. Therefore more investigations are required to shed more light on the miR-802 regulatory network and its potential prognostic value 27.
In the past several years, bioinformatic analysis of large-scale gene expression and clinical data have been an effective and applicable tool for investigating signaling pathways, hub genes, or tumorigenesis mechanisms in different cancers 29-31. Analyzing biological data by using computational and bioinformatic tools and websites is a useful approach for discovering new biomarkers that could have potential importance in clinical research and routine 32. Determining prognostic biomarkers based on bioinformatic approaches is among the recent interesting investigation areas and is performed for different types of cancers 33-35.
In this study, we explored the miR-802 target genes, signaling pathways protein-protein interaction network, and key cluster and hub genes via bioinformatics tools. Then the resulted hub genes were assessed to predict their potential biomarker value for prognosis in breast cancer. Figure 1 shows the workflow of the present study.
Materials and Methods :
Evaluation of the prognostic value of miR-802
Pan-cancer survival analysis of miR-802 in TCGA
dataset: Analysis of pan-cancer overall survival, utilizing pan-cancer TCGA miRNA database was performed by Kaplan-Meier Plotter (HTTP:// kmplot.com/analy-sis/) 36,37. Kaplan Meier plotter performs its assessments based on mRNA, miRNA, protein content of cancer samples. One of the sources of the Kaplan Meier plotter is the TCGA (The Cancer Genome Atlas) Dataset 38. TCGA contains a molecular characterization of over 20,000 primary cancerous and matched normal samples of 33 cancer types and has generated over 2.5 petabytes of genomic, epigenomic, transcriptomic, and proteomic (https://www.cancer.gov/tcga). We used the survival data of breast cancer (BRCA) tissues of 1078 patients from TCGA dataset (n=1078, BRCA). All molecular subtypes of breast cancers have been included and the overall survival of the patients was assessed. Split of the patients for low expression or high expression of miRNA was performed based on auto select best cutoff mode. In this way, all possible cut-off values between the lower and upper quartiles are computed and the best performing threshold is used. The false discovery rate of auto-selected cut-off was 1%. Log-rank p-value <0.05 is considered a statistically significant finding, and the hazard ratio is calculated with 95% confidence interval.
Integrated prediction of miR-802 target genes
We first used four highly used and well-known tools for the prediction of miR-802 target genes (miRDB: http://mirdb.org/, Targetscan7.2: http://www.target-scan.org/, MiRWalk 3.0: http://mirwalk.umm.uni/, and MiRWalk 2.0: http://zmf.umm.uni-heidelberg.de/mir-walk2). In the second step, we adopted an integrated approach to select target genes from mentioned databases. In order to get an insight of all possible molecular functions of a miRNA, the range of considered target genes should be comprehensive and wide enough. However, adopting a suitable approach would be challenging. Integrating the data of different research tools is becoming an interesting approach for the prediction of miRNA target genes 39,40. Due to large number of predicted genes in every database, while a target gene is annotated in more than one reliable database, it is usually a robust and valid one. In this regard we used TBtool software, a user-friendly toolkit, to analyze and visualize overlapping targets of miR-802 from four prediction data bases 41. Venn diagram and Upset Plot from TBtool were utilized to display the intersection of target genes between four microRNA predictive databases.
Gene ontology (GO) and kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analysis
The overlapping target genes selected by TBtool software were then analyzed by Metascape (metascape. org/gp/index.html) for GO and KEGG Pathway Enrichment Analysis. Metascape is a web-based portal that retrieves information from the latest version of databases and provides functional enrichment, gene annotation, interaction analysis, and so on to interpret OMICs-based studies 42. To GO and KEGG pathway enrichment analysis in this study, the names of the overlapped gene were inputted in Metascape online tool and the result was exported. GO analysis included Biological Processes (BP); Molecular Functions (MF) and Cellular Components (CC). p-value <0.05 was considered statistically significant for GO and KEGG pathway enrichment analysis. Transcription Factors (TFs) related to target genes were retrieved from TRUST (version 2.0) database which contains the data of regulatory relationship of Transcription factors 43. TRUST analysis was performed on terms with a p-value <0.01 which were collected and grouped into clusters. The trust data is accessible from Metascape as well.
Protein-protein interaction (PPI) network
STRING database (STRING, https://string-db.org/) version 11 was used to analyze the Protein-Protein Interaction (PPI) of overlapping genes. A confidence score higher than 0.4 was set to build the interaction network. The PPI network of genes was then extracted for more analysis by Cytoscape software (version 3.7.1). Potential clusters and modules of the PPI network were screened by Molecular Complex Detection (MCODE) plugin Cytoscape. MCODE finds highly interconnected regions in PPI network. Detected modules with MCODE score >3 and nodes number >2 were presented.
Analyzing the overall survival rate and expression of target genes
To analyze the potential prognostic value of selected target genes obtained from GO and network analysis, overall survival analysis was carried out by submitting the selected gene names in the Kaplan-Meier Plotter mRNA data set (p-value adjustment, 0.05 significance). Expression analysis of the selected genes was performed by the GEPIA database as well. p-value <0.05 was considered significant for mean expression differences between normal and tumor samples of breast cancers. GEPIA database used the expression data of 1085 tumor samples and 291 normal samples of breast cancer patients retrieved from TCGA.
Results :
Prognostic values of miR-802 in breast cancer and in silico exploration of its target genes: To evaluate the
prognostic values of miR-802 in breast cancers, a breast-cancer survival analysis was performed based on the breast-cancer TCGA miRNA database. The results showed that the high expression of miR-802 was significantly correlated with poor patient survival rate (BC, p=2.7e-5) which demonstrates its biological importance (Figure 2). Based on this analysis, miR-802 and its target genes might have the potential to be considered as a biomarker for breast cancer prognosis.
To figure out more about the roles of miR-802 in the biological processes of cells and its regulatory network, the predicted targets of miR-802 were retrieved from miRDB, Targetscan, miRwalk 2, and miRwalk 3. A set of genes was predicted by every database that intersection genes of these target genes were visualized by TBtool software in figure 3A (Figure 3A). This software perfectly provides clusters between databases and each cluster gene can be easily extracted from software. As is displayed in figure 3A, many genes have been annotated in more than one database. Prediction of a specific gene in different databases illustrated the higher probability of that gene to be the accurate target of miR-802. Therefore we selected overlapping genes, and in order not to miss any possible information, we chose every overlapping gene between two databases and more. In this way, 247 genes were selected for further analyses which are shown in figure 3B (Figure 3B).
Functional analyses of the predicted target genes
For GO annotation and KEGG Enrichment analyses of 247 selected target genes of miR-802, the Metascape which is an online functional enrichment tool, was exploited. p-values <0.05 were considered significant for enriched terms. Figure 4 shows the result of the molecular function gene ontology of 247 target genes. For GO enrichment analysis on the MF level, the selected target genes were mainly enriched in transcription coregulator activity, kinase regulator activity, chromatin binding and mRNA binding. On the BP level, the genes were mainly enriched in embryonic morphogenesis, regulation of cell cycle process, regulation of protein kinase activity, transmembrane receptor protein tyrosine kinase signaling pathway, regulation of cellular protein localization and Wnt signaling pathway. On the CC level, the genes were mainly enriched in axon,
dendrite, glutamatergic synapse, spliceosomal complex and cytoplasmic ribonucleoprotein granule. Other significantly enriched GO terms are represented in table 1.
The most significant result of GO and KEGG pathway enrichment along with two other databases has been Wnt signaling pathway, foxo signaling pathway, Pathways Affected in Adenoid Cystic Carcinoma, and regulation of dephosphorylation (Table 1). Analysis of TFs by TRUST showed that RUNX3, FOXO3, and E2F1 are possible TFs that regulate miR-802 expression and its target genes network (Figure 5).
PPI network construction and survival analysis of clustered genes
247 overlapping target genes included in PPI Network exploration via the STRING database with a medium confidence score (interaction score >0.400). The resulted network is displayed in figure 6 which has 247 nodes and 366 edges. PPI enrichment p-value is 6.46e-10. Three clusters with highly interconnected regions were extracted from PPI network by the MCODE clustering plug-in with MCODE score >3 and nodes number >2. Highly connected genes in a network mostly played an important role in the biological processes and would be taken as hub genes. 21 genes were involved in the currents study resulted clusters and considered as hub genes. Figure 7 shows the three selected clusters and their involved genes. It is supposed that these 21 genes might have an important function in miR-802 molecular processes and regulatory networks. Therefore the prognostic value of them in breast cancer was evaluated by Kaplan-Meier plotter (http://kmplot. com/analysis/) on gene expression data of 1880 breast cancer patients. The result show that among these 21 genes, high expression of CASC3 (207842_s_at), ITGA4 (213416_at), AGO3 (EIF2C3/219426_at), TARDBP (221264 _s_at), MED13L (THRAP2/ 212209_at) and SF1 (ZNF162/208313_s_at) genes are positively correlated with patient survival, while high expression of SNRPE (203316 _s_at) and CRNKL1 (219913_s_at) indicate poor patients survival (Figure 8). It seems these 8 genes could be considered and experimentally evaluated as a prognostic biomarker for breast cancer. Moreover, expression analysis in the GEPIA database showed that the mean expression of 12 genes (CASC3, MED13L, SF1, CRNKL1, SNRPE, SYNCRIP, RBM8A, RAN, CIT, LUC7-L3, WBP11, KPNA1) out of 21 selected genes, is significantly (p-value <0.05) different between normal and tumor breast samples, and 5 genes with prognostic value are among differentially expressed genes as well
(Figure 9). The differential expression pattern of most of miR-802 hub genes in breast cancer samples, made the probable role of this microRNA in breast cancer development stronger.
Discussion :
MicroRNAs play several key regulatory roles in biological processes 44. Similarly, miR-802 has shown multi functions in different conditions. It had been reported that miR-802 is involved in several malignancies such as liver cancer 22, prostate cancer 45, gastric cancer 23, cervical cancer 24, ovarian cancer 46, pancre-atic cancer 47, and so on. Other studies had shown the association of miR-802 with impaired glucose metabolism and obesity 48,49. There were also some reports of physiological functions of miR-802 in kidney or intestine development 50,51. In consistent with previous studies, the in silico investigation of the current study showed similar results that would be argued in the following. Deciphering miR-802 target genes in the large picture, we applied an integrated approach for target prediction from four miRNA-Target gene prediction tools that resulted in 247 selected genes. The most significant GO terms that was enriched from these selected genes, were Wnt signaling pathway (hsa04310, p-value: -6.25), regulation of cell cycle process morphogenesis (GO:0010564, p-value: -4.64), and FOXO pathway (hsa04068, p-value: -4.54) which all are among the molecular mechanisms of cancer development that suggest the potential role of miR802 in cancer progression and probable pathways that this micro RNA plays role in them 52-54. In addition to our results, there are other evidences that support the role of miR-802 in Wnt signaling pathway which verify the reliability of our results. It is reported that miR-802 and miR-1 regulate mesenchymal-epithelial transition during kidney development by regulation of Wnt-4/β-catenin signaling 55. In addition, it has been shown that Tmed9, a modulator of Wnt and lysozyme/defensin secretion in the mouse small intestine, as well as Fzd5 and Tcf4, the downstream components of Wnt signaling, are targeted and suppressed by miR-802 56.
As illustrated in the results, E2F1was identified as one of the transcription factors that regulate miR-802 expression. The important issue that should be mentioned here is the known regulatory effect of E2F1 on several genes involved in cell cycle which is verified in different literature 57-59. On the other hand in silico findings of the current study demonstrated that miR-802 played role in the cell cycle process (GO:0010564), mitotic cell cycle phase transition (GO:0044772) and cell division (GO:0051301). Consistent with our results, there was also a report on effect of miR-802 in inhibiting cancer cell proliferation by targeting FOXM1 and suppressing Cyclin A and Cyclin B1 which are key regulators of cell‑cycle progression 27. Taken together, it seems that miR-802 is an effective microRNA in the cell cycle that its regulatory network could be further explored.
FOXO (Forkhead box O) is a transcription factor that plays important role in fundamental cellular processes 60. The result of this study indicated that FOXO is another transcription factor of miR-802 and demonstrated its role in FOXO signaling pathways (has-04068). Consistent with our result, it is reported in another study that FOXO regulates transcription of miR-802 which supports our finding 49.
Among other terms that are shown in figure 4, were transcriptional regulation of white adipocyte differentiation (R-HAS-381340) and regulation of dephosphorylation (GO:0035303); both of which are involved in cellular metabolisms 61-63. In line with our results, it has been demonstrated that dysregulated miR-802 is critically associated with glucose metabolisms, obesity and some of their most important related conditions 48,64. Hence, by considering the results of the bioinformatics ontology study of miR-802 target genes and the consistency of the results with the literature, it could be validated that the platform of the study and integrated target prediction was promising and applicable approach.
Regarding the prognosis value of candidate genes, our result using bioinformatics tools demonstrated that CASC3, ITGA4, AGO3, TARDBP, MED13L, SF1, SNRPE and CRNKL1 could have prognostic value in breast cancer. Supporting our findings, the prognostic value of most of these genes has been reported in breast cancer or other malignancies, ether in silico or experimental. In agreement with our result, another study on the RNA network of triple negative breast cancer has verified that TARDBP had prognostic value 65. Furthermore, another in silico study suggested the prognostic value of ITGA4 in basal-like and HER2+ breast cancer 66. In an integrated study utilizing public bioinformatics datasets, ITGA4 has been identified as a prognostic marker of early ovarian cancer 67. Other studies also demonstrated a significant association of SF1 with the overall survival of pancreatic cancer through aberrant alternative splicing events 68. Moreover, SF1 regulated interferon l which had prognostic value in many cancers 69. In lung cancer, SNRPE and MED13L had been reported as potential prognostic markers 70,71. Furthermore, it has been indicated that CASC3, AGO3 and TARDBP could have a significant role in the prognosis of gastric cancer, hepatocellular carcinoma and cervical cancer respectively 72-74.
Regarding 21 selected target genes of miR-802 in PPI clusters, there are several articles indicating that almost all of these genes are involved in different malignancies including breast cancer. Some of the genes that have evidence in breast cancer are DDX21 which regulates epithelial-mesenchymal transition in breast cancer 75, LUC7L3 inhibits breast cancer progression 76, CDK19 plays regulatory function in triple-negative breast cancer 77, downregulation of Ran GTPase limits proliferation and migration of breast cancer cells 78, KPNA1 involves in tamoxifen resistance 79, DDX3X affects breast cancer cell cycle 80 and so on.
About molecular function of selected genes in other cancers we can mention, WBP11 that inhibits gastric cancer migration 81, AGO3 that regulates the Wnt/β-catenin signaling pathway in cervical cancer 82, MED13L which is functional in lung cancer radiosensitivity 71, SYNCRIP that is involved in poor prognosis of pancreatic cancer 83 and Down-regulation of CIT which limits human bladder cancer cells proliferation 84. All of these make the possible role of miR-802 in breast cancer more strong, therefore the regulatory network and prognostic value of its target genes can be further studied in breast cancer.
Conclusion :
This study provides comprehensive insight into miR-802 functional roles and regulatory networks. Besides the survival analysis provides promising candidate targets for the prognosis of breast cancer. Additionally, it is demonstrated that the exploitation of bioinformatics tools could be a reliable and effective approach in medical investigations.
Acknowledgement :
This work was supported by Iran University of Medical Sciences (IUMS), Tehran, Iran under Grant number 16162. This manuscript was extracted from the Ph.D. thesis of Maryam Eini and was supported by Project number (16162) from the Vice-Chancellor for Research Affairs of Iran University of Medical Sciences. We thank our colleagues at the Department of Biotechnology, Faculty of Allied Medicine, the Iran University of Medical Science for their supportive discussions.
Funding: This work was supported by Iran University of Medical Sciences (IUMS), Tehran, Iran under Grant number 16162.
Conflict of Interest :
The authors declare that they have no competing interests.
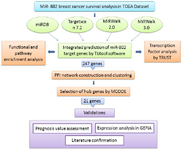
Figure 1. The workflow of the current study. This figure is a graphical overview of datasets, software and bioinformatic analysis that was exploited to investigate miR-802 target genes.
|
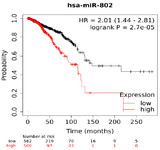
Figure 2. Overall Survival (OS) of miR-802 based on TCGA miRNA in breast cancer.
|
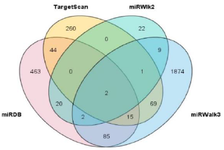
Figure 3A. Venn diagram shows the intersection of target genes that were retrieved from four miRNA target prediction tools (miRDB, TrgetScan 7.2, mirWalk2, and mirWalk3).
|
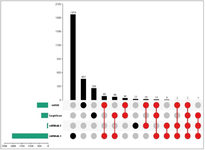
Figure 3B. Upset Plot displayed Overlapped target genes between four databases which were 247 genes in total and are marked in red.
|
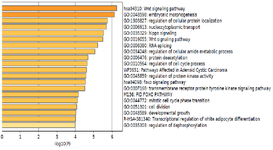
Figure 4. Bar chart of enriched terms resulted from gene ontology analysis.
|
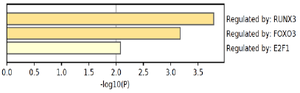
Figure 5. predicted transcription factors that regulate miR-802 target gene network.
|
Figure 6. The PPI network of 247 selected target genes of miR-802. Nodes and edges represent genes and interactions between them respectively.
|
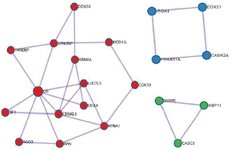
Figure 7. Clusters extracted from PPI network by MCODE. Three clusters and their involved genes are shown in different colors.
|

Figure 8. Prognostic value of miR-802 target genes. These 8 genes have better prognosis in breast cancer patients.
|

Figure 9. Gene expression analysis of selected hub genes. The red color bar represents gene expression in tumor samples (n=1085) and the gray color bar represents gene expression in normal samples (n=291). BRCA: breast cancer, (p-value <0.05).
|

Table 1. Summary of pathway and GO enrichment analysis
The count stands for the number of miR-802 target genes which are involved in the enriched term.
% stands for percent of gene per enriched term.
Log10(P): stands for the p-value regarding each term enrichment.
|
|