Exploring Potential Biomarkers Underlying Pathogenesis of Alzheimer’s Disease by Differential Co-expression Analysis
-
 Izadi, Fereshteh
Department of Genetics, Evolution and Environment, Darwin Building, University College London (UCL), Gower Street, London WC1E 6BT, UK, Tel: +44 7846280861, E-mail: izadi1991@yahoo.com
Izadi, Fereshteh
Department of Genetics, Evolution and Environment, Darwin Building, University College London (UCL), Gower Street, London WC1E 6BT, UK, Tel: +44 7846280861, E-mail: izadi1991@yahoo.com
-
Department of Genetics, Evolution and Environment, Darwin Building, University College London (UCL), London, UK
Abstract: Background: Alzheimer's Disease (AD) is the most common form of dementia in the elderly. Due to the facts that biological causes of AD are complex in addition to increasing rates of AD worldwide, a deeper understanding of AD etiology is required for AD treatment and diagnosis.
Methods: To identify molecular pathological alterations in AD brains, GSE36980 series containing microarray data samples from temporal cortex, frontal cortex and hippocampus were downloaded from Gene Expression Omnibus (GEO) database and valid gene symbols were subjected to building a gene co-expression network by a bioinformatics tool known as differential regulation from differential co-expression (DCGL) software package. Then, a network-driven integrative analysis was performed to find significant genes and underlying biological terms.
Results: A total of 17088 unique genes were parsed into three independent differential co-expression networks. As a result, a small number of differentially co-regulated genes mostly in frontal and hippocampus lobs were detected as potential biomarkers related to AD brains. Ultimately differentially co-regulated genes were enriched in biological terms including response to lipid and fatty acid and pathways mainly signaling pathway such as G-protein signaling pathway and glutamate receptor groups II and III. By conducting co-expression analysis, our study identified multiple genes that may play an important role in the pathogenesis of AD.
Conclusion: The study aimed to provide a systematic understanding of the potential relationships among these genes and it is hoped that it could aid in AD biomarker discovery.
Introduction :
Aging causes an increasing susceptibility to cognitive performances due to a developing neurodegeneration leading to neurologic disorders, such as dementia. More than 20 million people worldwide suffer from dementia, and this number is expected to exceed 80 million by 2040 because of the rapid increase in the numbers of the elderly 1. Alzheimer’s Disease (AD) is an irreversible progressive neurodegenerative disease affecting the central nervous system. Despite the increasing rate of AD incidence, no therapeutic strategy has been developed yet 2. Pathophysiologically, AD-related brain severe shrinkage caused neural and synaptic degenerations 3. The mentioned degenerative events can be detected in post mortem examination of patients suffering from severe memory loss 4,5. It is thought that the loss of memory is because of aggregating beta amyloid (Aβ) and Neurofibrillary Tangles (NFTs) of hyper-phosphorylated tau protein 1,6. Additionally, inflammation characterized by activated microglia 7 and oxidative stress, which result from an imbalance of Reactive Oxygen Species (ROS) and antioxidants 8,9 were shown to be associated with AD. Epigenetic changes happening in pre-frontal region by aging were shown to be related with AD functioning at cognitive level 1.
Rewiring of the biological networks to detect co-regulated and co-expressed units will help to facilitate looking into network’s components and depicting the relationships between interconnected genes. Gene co-expression networks enable us to highlight molecular mechanisms underlying diseases 10 and are considered as one way to investigate the etiology of AD efficiently. A large number of co-expression network methods have been proposed in the literature 11,12. Differential Co-expression Analysis (DCEA) offers a powerful approach for exploring phenotypic changes 13. Not only is AD etiology incompletely understood but also differences at transcriptome level and the genes potentially related to each distinct regions of brain are not recognized causing AD to be remained somewhat unclear. In the present study, a high-throughput genomic screening approach was applied using DCGL software and comparative microarray analyses. It was hypothesized that the distinct transcriptional changes in different regions of brain lead to AD-associated brain damages. Therefore, the transcriptional profiles from the gray matter of frontal and temporal cortices were compared with hippocampi derived postmortem brains to dissect AD pathogenesis in these areas. The rationale behind the used network approach is to prioritize AD-causative genes that are apart from differential alterations in their expression and are differentially regulated by Transcription Factors (TFs) between contrasting samples. For this, Differential Regulation Analysis (DRA) has been conducted on three separated regions of AD brains as contrasting samples.
Materials and Methods :
Data acquisition and pre-processing: The CEL files for GSE36980 series were downloaded from the GEO (http://www.ncbi.nlm.nih.gov/geo/) database and normalized with RMA method by using the Linear Models for Microarray Data (limma) R package. The main reason for selecting and exploiting this dataset is that GSE36980 series cover interspecies transcriptome analysis of various regions in gray matter in postmortem brains suiting the goal of dissecting pathological alterations in AD in several brain areas. Moreover, a number of researches have previously used these series and therefore would be able to compare the findings. After removing ambiguous probes, the extracted probe IDs were transformed into gene symbols. This data consists of a total of 79 samples (Table 1) based on the platform of GPL6244 and correspond to the frontal and temporal cortices and hippocampus.
Network construction: The DCGL R package was used to conduct DCEA 13,14. This software firstly calculates Differential Co-expression profile (DCp) and Differential Co-expression enrichment (DCe) to extract significant co-expression changes between a pair of genes in control and treatment samples. Next, Differentially Co-expressed Genes (DCGs) and Differentially Co-expressed Links (DCLs) were summarized from DCp and DCe values.
Next, DCGs and DCLs were extracted from DCp and DCe values previously calculated by DCp and DCe functions. DCp filters co-expression values of a pair of genes were assessed in control and treatment conditions. X and Y were defined as a subset of the gene pairs, where n is co-expression neighbors for a gene;
X = (xi1, xi2, …, xin)
Y = (yi1, yi2, …, yin)
The DC of a given gene is calculated with the following equation:
If the resulting DCGs and DCLs coincide with a TF, they will be referred to as a DRG and DRL, respectively. The DRGs and DRLs were scrutinized by DRsort function in Differential Regulation Analysis (DRA) module. In fact, DRA module identifies potential TF as upstream regulators of DCGs and DCLs 13. Finally, for illustrating the interactions between DRGs and their regulators, a network of DRGs and coincided TFs obtained by DRA was built for each of the datasets. By utilizing the Network Analyzer 15 nodes were set within networks with higher connections to darker color and bigger size.
Gene ontology, pathway analysis and visualization: To find the significantly over-represented biological GO terms and functions of gene products within a co-expression network of DRGs and DRLs, functional classification was performed using BINGO Cytoscape plugin 16 running hypergeometric test and Benjamini & Hochberg FDR correction at significant level 0.01. Finally, the clusters were visualized by Enrichment map Cytoscape plugin with Jaccard’s coefficient 0.001. DRGs were further functionally classified by PANTHER database (http://pantherdb.org/) to underlying pathways (Figure 1).
Results :
Co-expression analysis: The expression values of GSE36980 datasets were analyzed by utilizing DCGL v2.0 R package with default parameters. A total of 17088 unique genes were subjected to expression based filter and variance based filter, two functions embedded in DCGL to filter out genes that expressed extreme invariability across control and AD samples yielding 8544 and 2918 genes, respectively (Supplementary file 1). Afterward, using 2918 unique genes, co-expression analysis was performed on temporal cortex, frontal cortex, and hippocampus datasets separately. Expression based filter removes genes whose mean expression between experiments is lower than the median of this value for all genes and variance based filter removes genes that are not significantly variable than the median gene 13. In order to prioritize seed genes which are potentially related to AD pathogenesis, common and significant DRGs were selected using Targets’ Enrichment Density (TED) analysis and Targets’ DCL Density (TDD) analysis. TED and TDD identify differential co-expression genes and link in a particular TF’s targets, respectively 13. To this end, targets of significant TFs were extracted from 19,9950 TF-to-target interaction pairs as a library in DCGL v2.0 software 13. These pairs were further filtered out based on DRLs. In sum, 7, 19 and 13 genes were identified in temporal cortex, frontal cortex and hippocampus, respectively (Table 2). Significant TFs derived by TED and TDD analysis were used to infer co-expression network of DRGs in each dataset independently (Figure 2, Supplementary figures). DRGs were classified in terms of response to lipid, response to fatty acid, regulation of transcription from RNA polymerase II promoters and regulation of nitrogen compound metabolic process (Figure 3). Moreover, in pathway analysis, signaling pathways such as glutamate and G-protein signaling pathways were noteworthy (Figure 1).
Temporal cortex: 460 DCGs and 33656 DCLs were summarized using DC sum function to a final set of DCGs and DCLs (Supplementary file 2). There were 199 significant TFs in the results of TED analysis and 35 significant TFs in TDD analysis. 35 TFs that were significant in both of these two analysis results were chosen (Supplementary file 2). DRA analysis yielded 7 DRGs and 33 DRLs. DRGs were not only differentially co-expressed but also differentially co-regulated with 35 mentioned TFs. Then, a network of DRGs and DRLs was visualized using Cytoscape 3.4.0. Based on figure 2, PAX5 transcription factor and genes including ARID1A, CDC42 and LPPR4 were highlighted as the most important units within the genes network with more interconnected links (Figure 2).
Frontal cortex: In frontal cortex datasets, 628 DCGs and 166256 DCLs were summarized to 20 DRGs and 164 DRLs (Supplementary file 3). There were 199 significant TFs in TED analysis result and 135 significant TFs in TDD analysis result from which 135 TFs were chosen that were significant in both TED and TDD results (Supplementary file 3). In the inferred network, PAX5 and IKZF1 as TFs and genes including GRIK3, MAGI3, PRRX1 and DCAF6 were found as highlighted nodes with more connectivity.
Hippocampus: According to hippocampus datasets, 670 DCGs and 56264 DCLs were summarized to 16 DRGs and 43 DRLs (Supplementary file 4). There were 199 common and significant TFs in TED and TDD analysis which were used for inferring differential co-expression network with DRGs. There was more connectivity in hippocampus network than the other two networks. PAX5, ARNT, GATA1, EGR3 and IKZF1 TFs and genes including KCNK1, CACHD1, FABP3 and CHRNB2 showed highlighted roles as network nodes.
Discussion :
Aging is believed to be one of the most important non-modifiable risk factors of cognitive diseases that lead unequivocally to a number of detrimental changes in the neural system, increasing neuromorbidity and mortality. AD, as a progressive neurodegenerative disorder with no effective treatment options, is typically characterized by the presence of amyloid-beta plaques and hyper phosphorylated paired helical filament tau protein-rich neurofibrillary tangles 1. The identification of co-expressed genes related to AD presumably provides insights into the underlying mechanisms; in other words, a combination of gene effects likely holds promise as a more effective approach for detecting disease associated genes 42. In fact, examining co-expressed genes in spite of the individual genes could be more informative to explore genes that cause mental health disorders, such as AD 43,44. In this case, the correlation between two genes varies in distinct samples and thereby they are referred to as being differentially co-expressed. This correlation may change independently from the expression levels of two genes, indicating that transcriptome analysis merely based on differential expression analysis could miss important clues of regulatory patterns 45. Co-expression analysis has been performed for deciphering molecular mechanisms underlying mental health disorders 46-49. In the context of a well-established network analysis approach and given the most variable transcripts between control and AD brain samples, attempts were made by DCGL framework to explore putative pivotal genes that may be associated with AD. This work attempted to identify DRGs and links DRLs in AD by comparing expression datasets of temporal and frontal cortices and hippocampi. A comprehensive search in the literature showed that the obtained DRGs of AD brains mostly have direct or indirect links with AD or another neurologic disorder (Table 2). They are implicated in the gene ontology terms and shared biological pathways like response to lipid, fatty acid, nitrogen compound metabolic process and glutamate signaling pathways (Figures 1 and 2, supplementary file 5). Reportedly, considering GO terms as the response to lipid and fatty acid, brain lipid homeostasis plays an important role in AD 50. In this regard, differential regulation of delta 4-desaturase, sphingolipid 1 (DEGS1) and fatty acid binding protein 3 (FABP3) in hippocampus and lipid phosphate phosphatase-related protein type 4 (LPPR4) in temporal cortex datasets may fairly explain the relationship between brain damages happening in these regions and lipid metabolism. DEGS1 encodes a member of the membrane fatty acid desaturase family which is shown to interfere in AD via lipid rafts 25. FABP proteins are thought to participate in the uptake, intracellular metabolism and/or transport of long-chain fatty acids. Concordantly, serum levels of brain-type FABP are elevated in a significant proportion of patients with various neurodegenerative diseases including AD 38. LPPR4 acts as phospholipid dephosphorylate involving axonogenesis. The control of ion flow across the lipid membrane is essential for many cellular functions, including neuronal excitability and dysfunction of conveying ions through lipid bilayers involved in multiple neurologic diseases 51. As illustrated in figure 1, the DRGs are more implicated in signaling pathways; but the DRGs from frontal cortex were more enriched in ionotropic glutamate receptor pathway and metabotropic glutamate receptor group II and III pathways. The dysregulation of glutamatergic signaling has been shown to be associated with AD. Glutamate acts via ionotropic glutamate receptors (iGluR) and metabotropic glutamate receptors (mGluR), both of which have been implicated in AD 52. Differential regulation of glutamate receptor ionotropic, kainate 3 (GRIK3) and voltage-dependent R-type calcium channel subunit alpha-1E (CACNA1E) in frontal cortex datasets may be biologically relevant with the mentioned pathways in AD brain areas. Concordantly, a significant change in the expression of the GRIK3 gene was detected in a patient diagnosed with severe developmental delay 53. Many different kinds of signaling pathways are changed in AD, indeed the relevance of the biological pathways shown in figure 1 such as cytoskeletal regulation by Rho GTPase suggests mediating of these signaling pathways in the different lobs of brain, in this case in temporal cortex with differential regulation of CDC42. CDC42 has been linked to neuronal diseases like Alzheimer and Parkinson's disease through its role in cytoskeletal organization 54. Among the DGRs, CNTN2, KCNK1 and S100A1 were found common in frontal cortex and hippocampus datasets. S100A1 encodes for calmodulin signaling molecules. Increased levels of calmodulin have been reported in the hippocampus of AD model mice 55. These changes seemingly show an aberrant involvement of calmodulin in the impairment of cell cycle control in AD. As for the potassium channel subfamily K member 1- KCNK1, recent genetic studies suggest a central role for neuroinflammation. KCNK1 is a voltage-gated potassium channel upregulated by activated microglia and a mediator in amyloid-mediated microglial priming, additionally reactive oxygen species production that was shown to be related with autoimmunity 56. CNTN2 has been shown to undergo nuclear translocation and altered transcription 33.
These findings probably show that hippocampus and frontal cortices might deeply play a role in AD by mediating with conveying ions. Their obtained DRGs participated in vital processes like signaling, ion transportation and homeostasis. However, these processes mostly signal pathways somehow shared with temporal cortex implying the role of signal molecules within and between brain areas in neurologic dysfunctions. Concordantly, a comprehensive study has been already carried out on GSE36980 series to examine the alteration in the expression of diabetes-related genes in AD brains where they illustrated that hippocampi of AD brains have the most significant alteration in gene expression profile 57.
With a glance at table 2 and the terms including amyloids, inflammation, ROS and immune system, one could infer a cascade of events in which the DRGs interfere. Beta-amyloid deposition following the activation of microglia will initiate an inflammatory response leading to the release of potentially neurotoxic substances and ROS that targets neural damage 58. Afterward, along with immune response, nitrogen compounds will mediate to reverse the consequences of oxidative stress in damaged regions 8,9. In sum, it was shown that DRGs covered a wide range of known functions and processes implicated in main AD signaling pathways. In a study by Satoh et al 59, GSE36980 series used in the present study were utilized to identify biomarker genes relevant to the molecular pathogenesis of AD. They analyzed a RNA-Seq dataset composed of the transcriptome of postmortem AD brains derived from two independent cohorts and they identified the core set of 522 genes deregulated in AD brains shared between both, compared with normal control subjects. Notably, in agreement with our study, LPPR4 was bolded in AD brains in both microarray and RNA-seq datasets. By consistent downregulation of NeuroD6 in AD brains, the results indicated that downregulation of NeuroD6 serves as a possible biomarker for AD brains. Previous studies identified LPPR4 as direct target genes for NeuroD6 by binding assay to E-boxes located in target gene promoters 60. GSE36980 series were also employed by Fowler et al 61 used to investigate potential underlying biology in AD and in concordance with the results of the present study, they noticed the over-representation of glutamate in their data mining. They first identified genes consistently associated with AD in each of the four separate expression studies, and confirmed the result using a fifth study. They next developed algorithms to search hundreds of thousands of GEO data sets, identifying a link between an AD-associated gene (NEUROD6) and gender. Additionally, they identified several genes related to glutamate (including CACNG3, a regulator of AMPA-sensitive glutamate receptors; SLC17A7, a mitochondrial oxoglutarate carrier; and GOT2, mitochondrial glutamic-oxaloacetic transaminase. In our study, differential regulation of glutamate receptor ionotropic, kainate 3 (GRIK3) and voltage-dependent R-type calcium channel subunit alpha-1E (CACNA1E) in frontal cortex datasets could be therefore biologically relevant with the mentioned pathways in AD brain areas. Moreover, in our study, differential regulation of Slc2a1 in hippocampus data seemingly implies the role of impairments in glutamatergic transmission mostly in hippocampus of AD brains. The role of glutamate transporters such as SLC1A6 was also highlighted in a study by Satoh et al 59.
Conclusion :
The purpose of the study was to explore the molecular mechanism in the development of AD, and a comparison of AD in three regions of the brain was done. Therefore, in the frame of network reconstruction and data mining approaches, a small number of possible genes and TFs were identified that their interplay could lead to neural dysfunctions toward AD. However, One should be cautious regarding small sample size while by utilizing more adequate samples, the results would be more reliable.
An expected outcome of such a work would possibly shed light on the bridges between AD-associated brain damage in transcriptome level and presenting crucial evidence in clinical diagnosis and treatment.
Acknowledgement :
We thank Dr. Antonio R. Franco of University of Cordoba (Spain) for his precious assistances.
Conflict of Interest :
The authors declared that they have no competing interests.

Figure 1. Bar chart of pathways potentially DRGs extracted from tempral cortex, frontal cortex and hippocampus expression data. PANTHER server with default parameters for pathway analysis was used for pathway analysis. The length of each bar showes how many genes have been assigned to a given pathway.
|
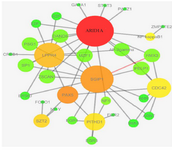
Figure 2. Differential co-expressed network of DRGs and DRLs captured by TED and TDD results in temporal cortex datasets. The bigger and darker nodes show the nodes with higher connectivity within the network.
|
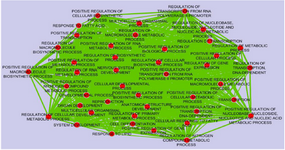
Figure 3. Functional classification of biological process in which Differential Regulated Genes (DRGs) were supposed to be involved. The GO terms were considered significant based on hypergeometric test with Benjamini & Hochberg FDR correction and significance level 0.01 by BINGO app. The results were illustrated using the Enrichment map Cytoscape 3.4.0 plugin. Ticker lines and bigger circles show more genes with higher significance level belonging to a given term.
|
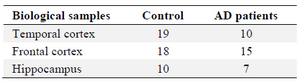
Table 1. Sample characteristics
|

Table 2. List of differential regulated genes (DRGs) and corresponding p-value<0.05 of differential co-expression enrichment (DCe) in temporal cortex, frontal cortex and hippocampus datasets
// showing DRGs with ambiguous role in neurologic disorders.
|
|