Construction of a High Efficiency PCR Products Cloning T Vector Using pGEM-5zf (+)
-
Zhao, Yaofeng
-
Division of Clinical Immunology, Department of Laboratory Medicine, Karolinska University Hospital Huddinge, SE-141 86 , Stockholm, Sweden
-
Liu, Zhancai
-
Department of Physics, Chemistry and Biology, Jiaozuo Teachers College, Jiaozuo, 454001 , Henan, P. R. China
-
Yu, Shuyang
-
Division of Clinical Immunology, Department of Laboratory Medicine, Karolinska University Hospital Huddinge, SE-141 86 , Stockholm, Sweden
-
Wen, Sicheng
-
Division of Clinical Immunology, Department of Laboratory Medicine, Karolinska University Hospital Huddinge, SE-141 86 , Stockholm, Sweden
-
Hammarstrom, Lennart
-
Division of Clinical Immunology, Department of Laboratory Medicine, Karolinska University Hospital Huddinge, SE-141 86 , Stockholm, Sweden
-
 Rabbani, Hodjattallah
Ph.D., Monoclonal Antibody Research Center, Avicenna Research Institute, ACECR, Tehran, Iran, P.O. Box: 19615-1177, Tel: +98 21 22432020, Fax: +98 21 22432021, E-mail: Hodjattallah.Rabbani@cck.ki.se
Rabbani, Hodjattallah
Ph.D., Monoclonal Antibody Research Center, Avicenna Research Institute, ACECR, Tehran, Iran, P.O. Box: 19615-1177, Tel: +98 21 22432020, Fax: +98 21 22432021, E-mail: Hodjattallah.Rabbani@cck.ki.se
-
Immune and Gene Therapy Laboratory, CCK, Karolinska University Hospital Solna , Stockholm, Sweden
-
Department of Antigen and Antibody Engineering, Monoclonal Antibody Research Center, Avicenna Research Institute, ACECR , Tehran, Iran
Abstract: A highly efficient cloning vector was constructed for cloning PCR products by inserting an 80 bp DNA fragment into pGEM-5zf (+) vector. The Xcm I digestion of this vector gave rise to a 3’ overhanging deoxythymidine offering the possibility of cloning PCR products with 3' adenosine overhang created by Taq DNA polymerase. Furthermore, two EcoR I sites were added to the construct for identification of recombinant plasmids using a single restriction enzyme. Taken together, the more efficient cloning performance and the lower cost of this vector as compared to the commercial T vector, suggests that it may be one of the best T vectors for cloning of PCR products.
Introduction :
Cloning of PCR products has become a standard routine in most molecular biological laboratories.
The commonly used cloning strategy utilizes the feature of Taq DNA polymerase to add a 3’ overhanging deoxyadenosine residues in most of the PCR products (1). Therefore, T vectors possessing two 3’ overhanging deoxythymidines have been created using different approaches, either by tailing a single deoxythymidine to the 3’ end of linear and blunt ended plasmids (2) or by cutting a modified plasmid with selected restriction enzymes such as Xcm I, to generate linear plasmids with single overhang deoxy-thymidine at both 3’ ends (3-8). Theoretically, the T vector prepared by utilizing the restriction enzymes should have a higher cloning efficiency than a tailed one. Very often, however, the plasmids are only partially digested by Xcm I (5,8), probably because the two Xcm I recognized sites are usually separated only by a few nucleotides, resulting in a high percentage of non recom-binant transformants. Several methods have been employed to circumvent this problem including dephosphorylation of linear T vectors (4) or introduction of a long spacer between the two Xcm I sites in order to make the fully digested vectors easy to recover and purify (6).
Furthermore, as the Xcm I recognizes 9 random nucleotides between the conserved CCA and TGG sequences, the efficiency of digestion could be increased by alteration of the recognition sequences even using a short spacer between the two Xcm I sites. In this paper, we have constructed a very effective T cloning vector based on this idea.
Materials and Methods :
An 80 base oligo (5’ gaagaggcggtcgacga-attcccaaaattaaaatggatcgatcgccatcgaaattttgggaattcactagtgtgccgtcc 3’), T-oligo, containing two Xcm I sites and two EcoR I sites flanked by Spe I and Sal I sites, was synthesized. To make a double stranded DNA (dsDNA), a PCR was carried out using the T-oligo as a template, and two primers, T-sense (5’ gaa gca gcg gtc gac gaa tt 3’) and T-antisense (5’ gga cgg cac act agt gaa tt 3’) and were designed based on the ends of the T-oligo.
The PCR conditions were 94?C/2 minutes, 30 cycles of 94?C /10 second, 58?C /15 second, 72?C /15 second, which yielded a specific amplification and no primer dimers could be detected after running the PCR products on an agarose gel.
The resulting PCR products were purified with a QIAGEN Gel Extraction Kit (QIAGEN, Valencia, CA, U.S.A), and subse-quently digested using Sal I and Spe I. The restriction reaction was cleaned using QIAGEN Nucleotide Removal Kit. The purified DNA was ligated with Sal I and Spe I double digested pGEM-5zf(+) and trans-formed into E.coli DH5? for screening.
Result :
As designed, insertion of the short oligo into pGEM-5zf(+) did not change the open reading frame of the LacZ gene and therefore the normal LacZ activity was maintained.
Approximately 90% of the blue clones were recombinants which contained an in-sertion with the expected sequence, while 10% were self-ligated plasmids, reflecting the fact that plasmids were probably cut only by either Spe I or Sal I. The recombinant plasmid with the correct insert was named pGEM-FT (Figure 1).
To test the cloning efficiency of the pGEM-FT vector, the plasmids were digested using Xcm I at 37?C overnight and separated on a 0.7% agarose gel. The linear plasmids were purified with QIAGEN Gel Extraction Kit (QIAGEN, Valencia, CA, U.S.A) and about 70 ng of the recovered vectors were ligated with 100 ng of an approximately 800 bp PCR product (rat Ig C? gene derived), produced by Taq DNA polymerase, using 3 Units of T4 DNA ligase. The ligation mixture was transformed into CaCl2 treated DH5? competent cells. Of a total of 803 transformed clones, 24 (approximately 3%) turned blue under color selection meaning that the pGEM-FT plasmid can be effectively digested by Xcm I. Most white clones were recombinants as proven by PCR (17/18), and the purified plasmids could be digested by EcoR I, liberating inserts with the expected size, indicating that the pGEM-FT vector digested by Xcm I could yield a high cloning efficiency, where more than 90% of trans-formants contain the correct insert.
A parallel experiment was also performed to compare the cloning efficacy of the pGEM-FT with the pGEM-T purchased from Promega (Madison, CA, U.S.A), where 60 ng of PCR products (263 bp, bovine IgE CH4 domain derived) were respectively ligated with 50 ng of both the vectors using 3U T4 DNA ligase. After transformation, pGEM-FT yielded totally 120 clones, of which 4 (3.3%) were blue, whereas the commercial pGEM-T yielded 14 blue clones out of 173 (8%), indicating the pGEM-FT produced a higher ratio of recombinants than the pGEM-T. The inserts could be easily released by EcoR I from the plasmids purified from all 10 randomly selected white colonies. Furthermore, sequencing analysis showed that the inserts were integrated into the pGEM-FT in the expected way (T/A cloning). However, more cloning experiments also showed that the longer the PCR product the lower the cloning efficacy of the PGEM-FT as seen with other vectors.
Discussion :
In summary, there are several advantages to the cloning of PCR products with pGEM-FT vectors. First of all, the pGEM-FT can be effectively double-digested by Xcm I to produce a highly efficient PCR cloning T vector, which offers up to 97% recombinant transformants under color selection. The high background of non-recombinant transfor-mants, which is frequently encountered using restriction endonuclease digested T vectors, is therefore largely reduced. Furthermore, the addition of two EcoR I sites to the plasmid allows the identification of recombinant plasmids using a single, cheap restriction enzyme. Taken together, the even better cloning performance and the lower cost of the pGEM-FT vector as compared to the commercial T vector, suggests that it may be one of the best T vectors for cloning of PCR products.
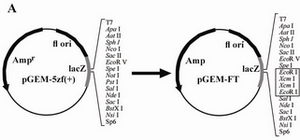
Figure 1. Construction of pGEM-FT using pGEM-5zf(+) (A) and the restriction sites of the short insert in pGEM-FT (B). The Spe I, EcoR I and Sal I sites are underlined. The Xcm I recognizing sequences are in upper cases. The arrows show the cutting sites of Xcm I. This insert was put into the pGEM-5zf (+) between the Spe I and Sal I sites.
|
|