Production of Pentameric Cholera Toxin B Subunit in Escherichia coli
-
Dakterzada, Farida
-
Department of Bacteriology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran
-
 Mohabati Mobarez, Ashraf
Department of Bacteriology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran, Tel: +98 21 82883862; Email: mmmobarez@modares.ac.ir
Mohabati Mobarez, Ashraf
Department of Bacteriology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran, Tel: +98 21 82883862; Email: mmmobarez@modares.ac.ir
-
Department of Bacteriology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran
-
Forouzandeh, Mehdi
-
Department of Biotechnology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran
Abstract: Cholera toxin B subunit (CTB) has been extensively studied as an immunogen, adjuvant, and inducer of oral tolerance in many investigations. Production of CTB has been carried out in the bacterial, plant, insect and yeast expression systems. In this study the expression of the CTB containing a 6XHis-tagged was performed by Escherichia coli (E.coli) M15. The yield of purified pentameric recombinant CTB was about 1 mg/l. Western blot analysis demonstrated that the recombinant CTB was antigenically active. In addition, GM1-ganglioside ELISA showed that recombinant CTB binds to GM1-gangelioside receptor, confirming disulfide bond formation and proper folding of the recombinant protein in E.coli. Overall, in regard to the vast applications of CTB in medicine, this bacterial expression system will be a fast, cost-effective and simple system for production of pentameric CTB and CTB conjugated proteins.
Introduction :
Vibrio cholerae (V.cholera) which causes cholera, a waterborne disease, affects thousands of life every year in developing countries (1). The symptoms of the disease are mainly caused by secretion of an enterotoxin named Cholera Toxin (CT). CT is composed of an enzymatic A (CTA) subunit and a cell binding B (CTB) subunit. The A subunit contains two non identical polypeptide chains, A1 (22 kDa) and A2 (5 kDa), linked by a single disulfide bond and is responsible for catalysis of adenylate cyclase to cause massive excretion of electrolytes from bowel, leading to the characteristic severe diarrhea of cholera. The B subunit of the toxin is composed of five identical, noncovalently associated polypeptide chains (11.6 kDa each), arranged in a ring-like pentameric configuration. The B subunit is responsible for the binding of the toxin to the cell by interacting with the hydrophilic carbohydrate moiety of the GM1 ganglioside present on most nucleated cells (2).
CTB is an important protein in medicine. It is a subunit vaccine candidate antigen for cholera (3,4). CTB is an effective mucosal adjuvant for oral and nasal vaccines. On the other word, when CTB is chemically or genetically conjugated to poor immunogenes it elicits serum and secretory antibodies against the fused antigens (5,6). CTB has also been used as a transmucosal carrier delivery system for induction of oral tolerance to conjugated autoantigens and allergens (7, 8). Up to now, CTB gene has been expressed in the bacterial (6,9,10), yeast (11), plant (12,13) and insect (14) expression systems. However, expression of recombinant proteins in some of these systems is limited by low accumulation levels and use of specific codons is needed (10,13). Condon modification of the genes could be a solution but it is laborious and expensive. It is noteworthy that quaternary structure is essential for CTB function, but this is impossible in some expression systems (9,11,15). The aim of this study was to produce native cholera toxin B subunit in its pentameric form in E.coli, which is a cost-effective and unsophisticated system for expression of proteins.
Materials and Methods :
Bacterial strains, vector and media: Hypertoxigenic V.cholerae strain 62013 was kindly provided by Pasture Institute of Iran, (Tehran, Iran). E.coli strains DH5α and M15 were obtained from Invitrogen (USA, Carlsbad), and Qiagen (USA, California), respectively. The pQE30 expression vector was purchased from Qiagen. Bacteria were cultured on LB agar or in LB and 2xYT broth (Merck, Germany) containing Ampicilin (100 µg/ml) and/or Kanamycin (25 µg/ml) (Sigma, USA).
Isolation and cloning of CTB: Genomic DNA from V.cholera strain 62013 was extracted using genomic DNA extraction kit (Bioneer, South Korea). Specific primers were designed according to ctxB sequence of the Vibrio phage CTX strain IB1482 (GenBank Accession No: GQ466612). A 309 bp coding sequence of ctxB was PCR amplified using specific primers. The forward primer was 5' CGCGGATCCACACCTCAAAATATTACTG 3' containing BamHI restriction site and reverse primer was 5' AATAAGCTTTTAATTTGCCATACTAATTGCG 3' containing HindIII restriction site. The reaction was carried out under the following condition: initial denaturation at 98°C for 4 min followed by 35 cycles of denaturation at 98°C for 10 sec, annealing at 55°C for 15 sec and extension at 72°C for 45 sec followed by a final extension time of 5 min at 72°C. The PCR product was analyzed by electrophoresis and the desired fragment was purified from agarose gel using high pure PCR purification kit (Roche, Germany). Following restriction digestion of the purified fragments with BamHI and Hind III enzymes, they were ligated to the similarly digested ends of pQE30 plasmid. DH5α E.coli competent cells were transformed with the ligation mixtures and the transformants were selected on LB agar plates containing 25 μg/ml kanamycin (16). Recombinant clones were analyzed by restriction analysis and PCR. Positive clones were further verified by DNA sequencing (Macrogen, South Korea).
Expression in E.coli: E.coli M15 was transformed with recombinant pQE30 plasmid. Some resistant colonies were inoculated in 5 ml of 2xYT medium supplemented with Ampicilin (100 µg/ml) and kanamycin (25 µg/ml) and grown overnight at 37C. The overnight cultivated recombinant bacteria were diluted 1:100 with 2xYT broth and subjected to further incubation. When OD600 reached 0.8, IPTG was added to the media at final concentration of 1 mM. Then, the incubation was continued at 37C for further 4 hr. The cells were then harvested by centrifugation and stored at −70C (16).
Purification and refolding of the recombinant protein: Purification was performed under hybrid condition (Invitrogen) with some modifications. Briefly, the cells were suspended in lysis/binding buffer (8 M urea, 20 mM Sodium Phosphate, 500 mM NaCl, pH=7.8) for 20 min at room temperature followed by centrifugation at 10000 rpm for 20 min. Then, the Ni2+-NTA resin column was equilibrated with the lysis/binding buffer and the supernatant was transferred to the column and mixed gently with resin by rocking the column for 1 hr at room temperature. The resin was then washed two times with denaturing wash buffer (8 M urea, 20 mM sodium phosphate, 500 mM NaCl, pH=6.0). Washing was continued by native wash buffer (20 mM sodium phosphate, 500 mM NaCl, 20 mM imidazole, pH=8.0) for two times. The recombinant protein was eluted in native elution buffer (20 mM sodium phosphate, 500 mM NaCl, 250 mM imidazole, pH=8.0) and finally dialyzed against Phosphate Buffered Saline (PBS), pH=7.4 to remove imidazole. Finally, to separate inactive monomeric from active pentameric forms of the purified CTB, one aliquot was subjected to an additional purification using Centricon 30 (Centricon, USA) according to instructions described by the manufacturer. The analysis was performed by 12% SDS-PAGE followed by comassie brilliant blue G-250 staining (Serva, Germany). The protein concentration was determined by Bradford method with bovine serum albumin as standard (Bio-Rad, USA).
Western blot analysis: The proteins separated by 12% SDS-PAGE were transferred onto polyvinylidene difluoride (PVDF) membrane (Roche, Germany) by using a semi dry blotter unit (Peqlab, Germany). The membrane was blocked by 3% skim milk in PBS. The blocked membrane was washed three times with PBS and incubated with 1:1000 dilutions of rabbit polyclonal anti CTB antibodies in 0.3% skim milk-PBST (0.1% Tween 20) for 1.5 hr. Then the membrane was washed three times during 30 min with PBST, and further incubated with a 1:5000 dilution of anti-rabbit IgG peroxidase conjugate (Abcam, UK) in PBST for 1 hr. Finally, the membrane was washed as described above, and developed with DAB reagent (Roche, Germany).
GM1-ganglioside binding assay: The affinity of rCTB protein for GM1-ganglioside receptor was determined by GM1-ELISA. A micro plate was coated with monoasialoganglioside-GM1 (Sigma, USA) (10 μg/ml in carbonate/bicarbonate buffer) by incubating at 4°C overnight. As a control, some wells were coated with BSA (10 μg/ml in carbonate/ bicarbonate buffer). Afterwards, the plate was washed three times with PBST (phosphate buffered saline containing 0.05% tween-20). The wells were blocked with 1% BSA in PBS at 37°C for 2 hr, followed by three washes with PBST. The wells were incubated with purified unboiled and boiled rCTB in PBS at 37°C for 2 hr (PBS alone was used as negative control) followed by washing three times with PBST. Then the plate was incubated with a 1:1000 dilution of rabbit polyclonal anti-CTB antibody at 37°C for 2 hr, followed by washing the wells three times with PBST. Then the plate was incubated with 1:5000 dilution of horseradish peroxidase conjugated anti-rabbit antibody (Abcam, UK) for 2 hr at 37°C and washed three times with PBST. Finally, the chromogenic substrate tetramethylbenzidine (Sigma, USA) was added to wells and the plate was incubated at room temperature for 30 min in a dark place for developing color, and then the reaction was stopped by the addition of 2N H2SO4. The optical density of the wells was read at 450 nm.
Results :
Isolation and cloning of ctxB: Based on Vibrio phage CTX strain IB1482, specific primers were designed to amplify coding sequence of ctxB. The expected size of PCR product, approximately 330 bp, was obtained following PCR (Figure 1A). Then, transformation of recombinant plasmids into the E.coli DH5α was performed and existence of ctxB in vector was confirmed by restriction enzyme analysis (Figure 1B). Finally, DNA sequencing confirmed the fidelity of the cloned sequence and its correct orientation in the plasmids (data not shown).
Expression and purification of recombinant protein: In the current study, we cloned the ctxB gene into pQE30 expression vector and used to transform the E.coli M15 expression host which not only CTB was expressed in this system, but also the expressed protein retained its pentameric conformation. The recombinant cells were harvested 1, 2 and 4 hr after induction, and the highest expression was achieved 4 hr after induction (Figure 2). The expressed recombinant protein was purified by affinity chromatography on a Ni2+-NTA column and refolded without any protein precipitation.
The purification and refolding of the protein was performed under hybrid conditions with some modifications. To monitor the formation of oligomeric CTB, the purified protein was re-suspended in sample buffer without β-mercaptoethanol and was directly loaded onto a 12% SDS-PAGE (Figure 2). Quantification of recovered protein yielded about 9 mg of the purified recombinant CTB per liter of induced culture. After separation of monomeric and pentameric molecules by membrane-filtration, the amount of pentameric CTB was estimated to be about 1 mg by the Bradford method. Finally, the rCTB was strongly and specifically recognized by rabbit polyclonal anti-CTB antibody which indicates its immune reactivity. Furthermore, western blot analysis under non-denaturing condition showed that the expressed CTB assembled into oligomers (Figure 3).
GM1-ganglioside ELISA binding assay: To confirm specific affinity of the pentameric rCTB, the GM1-ELISA method was carried out. The unboiled rCTB demonstrated a strong affinity for GM1-ganglioside (Figure 4), indicating that the expressed rCTB conserved its antigenic sites which are necessary for its binding to the GM1 pentasaccharide and also confirmed proper folding of the recombinant protein resulting in the functional pentameric structure. Almost no affinity was observed to BSA and PBS that were used as negative control for GM1 and CTB, respectively.
Discussion :
CTB is an important protein in medicine and has been extensively studied as immunogen, adjuvant, and inducer of oral tolerance in many investigations (3,7,17). Up to now, CTB has been expressed in several bacterial systems, such as E.coli (18), Lactobacillus (15), Bacillus brevis (19) and V.cholera (20). But expression of CTB gene in all of these systems often yield insufficient protein level and use of specific codons is needed (10,13). However, extensive codon modification of genes is laborious and expensive.
E.coli is a valuable organism for high-level expression of heterologous proteins. This is mainly because the bacterium is inexpensive to culture, and expression of recombinant proteins is comparatively fast. In this study we tried to express native rCTB in E.coli M15 expression system. Our results revealed that expression of rCTB in E.coli M15 system not only lead to high level expression of rCTB, but also analysis of purified protein with SDS-PAGE and western blotting under non-denaturing conditions demonstrated that this system is able to produce rCTB in its pentameric form. In addition, GM1-ganglioside ELISA binding assay demonstrated that the rCTB protein produced in E.coli represents a strong affinity to GM1 receptor. The presence of four vector derived amino acids and 6XHis tag at the N-terminal of rCTB resulted in a 13 kDa (instead of 11.6 kDa) protein and did not represent any apparent constraint for its refolding and activity. Our final recovered amount of functional protein was about 10% of the total expressed protein that is lower than the pentameric rCTB recovered by Areas et al (10), but is a great success in comparison to some other studies that could not produce pentameric form of this protein (9,11,15).
In purification method, we implied some modifications. We used urea instead of guanidine hydrochloride in lysis buffer. Efficacy of the lysis by urea was the same as guanidine hydrochloride and as lysis and binding buffers are the same one step was reduced in the purification process. Furthermore, application of high concentration (8 M) of urea can dissolve bacterial membrane and inclusion bodies. Therefore, by our method, we obviate isolation of inclusion bodies that is time-consuming and reduce the yield of purified protein considerably (21). We did not sonicate bacteria after lysis with buffer containing urea because the yield of purified rCTB with or without soniaction was the same.
Overall, here we present a very simple, rapid and cost effective procedure for purification of recombinant proteins that restore their quaternary structure and biological activity. Therefore, we suggest that this procedure should be tested for any particular recombinant protein. Recombinant CTB was immunologically characterized by Western blotting using polyclonal anti-CTB antibodies. The protein was strongly recognized by these antibodies.
Conclusion :
In conclusion, we hypothesize that this system will be an effective system for expression of other multimeric proteins and genetically conjugated proteins to CTB.
Acknowledgement :
This research was financially supported by Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran. We are grateful to Ali Jahanian Najafabadi for assisting in the English of the final draft. There was no conflict of interest.
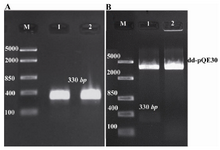
Figure 1. Agarose gel (1%w/v) electrophoresis. A) Isolation of ctxB by PCR. Lane 1 and 2: expected single bands of ctxB; Lane M: DNA size marker (Fermentase, Cat No: SM 1113). B) Digestion of recombinant vector by restriction endonucleases after plasmid extraction. Lane 1: recombinant vector, pQE30-ctxB, digested with BamHI and Hind III; Lane 2: pQE30 digested with the same enzymes. Lane M: DNA size marker (Fermentase, Cat No: SM 1113)
|
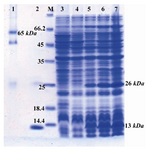
Figure 2. Detection of expressed and purified rCTB on SDS-PAGE (12% w/v) gel stained with coomassie blue G-250. Lane 1: purified rCTB under non-denaturing conditions; Lane 2: purified rCTB under denaturing conditions; Lane M: stand-ard protein size marker (kDa); Lane 3: pQE30 transformed bacteria; Lane 4: Un-induced sample; Lane 5, 6, and 7: in-duced bacteria after 1, 2, and 4 hr of induction with IPTG, respectively. In lane 2-7 presence of a band of about 26 kDa represents formation of dimeric CTB even after applying of denaturing conditions
|

Figure 3. Western blotting of recombinant CTB and its immunogenicity by polyclonal anti-CTB antibodies. Lane 1: purified CTB prepared under non-denaturing conditions; Lane 2: supernatant of pQE30 transformed bacteria; Lane 3: purified CTB prepared under denaturing conditions; Lane M: standard protein size marker (kDa)
|
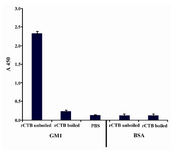
Figure 4. GM1-gangelioside binding assay of CTB pro-duced in E.coli M15. Wells coated with GM1 and BSA were incubated with boiled and unboiled CTB. PBS was used as negative control. The absorbance was measured at 450 nm
|
|