Generation of In-vitro Spermatogonial Stem Cells following Genetic Manipulation of Primordial Germ-like Cells
-
Mazaheri, Zohreh
-
Department of Anatomical Sciences, Faculty of Medical Sciences, Tarbiat Modares University , Tehran, Iran
-
 Movahedin, Mansoureh
Department of Anatomical Sciences, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran , Tel: +98 21 88011001; E-mail: mansoure@modares.ac.ir
Movahedin, Mansoureh
Department of Anatomical Sciences, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran , Tel: +98 21 88011001; E-mail: mansoure@modares.ac.ir
-
Department of Anatomical Sciences, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran
-
Rahbarizadeh, Fatemeh
-
Department of Biotechnology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran
-
Amanpour, Saied
-
Vali-e-Asr Reproductive Research Center, Imam Khomeini Hospital, Tehran University of Medical Sciences, Tehran, Iran
Abstract: Research about potential use of stem cells for the development of germ line cells in vitro had been challenged. In the present study, we reported a novel protocol consisting of cocktail growth factor addition for germ cell differentiation followed by transfection. The cells were purificated based on the expression on the cell surface of a protein. This protein is not present in normal cells of mice and does not interfere with cellular function. This cell surface marker is efficiently recognized by monoclonal antibodies. Bone marrow mesenchymal stem cells derived primordial germ like cells were differentiated to spermatogonial stem like cells by inducer cocktail including Retinoic acid (RA)+Leukemia inhibitory factor (LIF)+Basic fibroblast growth factor (bFgF). Co-culture system was used as a feeder under differentiated cells. A 400 bp fragment of spermatogonia-specific Stra-8 locus was enough to direct gene expression to the germ line stem cells. Stra8-CD4HAglo construct was used for purification of premeiotic differentiated cells. Expression of pluripotency (Pou5F1, Nanog, c-Myc) and specific germ cell )Mvh, Piwil2, Stra-8) genes in each stage were analyzed. The purified cells expressed the known molecular markers of PGC-like cells such as Mvh, Piwil2 & Stra-8. The outcomes of qPCR showed that ratio pluripotency of genes expression in selective group significantly decreased (p≤0.05) in the initial differentiation process. This results showed that ratio of Pou5F1, Nanog, c-Myc, Mvh, Piwil2 & Stra-8 expression to purified PGC-like cells were 0.41, 0.204, 1.1, 0.003, 0.184 and 2.276, respectively. Treatment of cells with RA affected up regulation of Stra-8. Although, c-Myc gene as an oncogenic gene had significantly increased (p≤0.05) at the end of differentiation stage compared to initial phase of study, this level of expression could not be tumorgenic. qPCR results of the differentiation stage showed higher expression of Stra-8 in co-culture+cocktail and co-culture groups, Also, there was a significant difference (p≤0.05) in the expression of Pou5F1 & Nanog. Our results suggest that selection and purification of PGC-like cells based on Stra-8 as a pre-meiotic marker is a useful tool for getting in vitro spermatogonial stem cell. This method facilitates identification of safely differentiated germ cells in vitro.
Introduction :
The development of Spermatogonia Stem Cells (SSCs) into gametes is an essential feature of sexual reproduction, although the development of SSCs and their differentiation into sperm is well understood on the genes controlling gamete formation, particularly during the early stages of SSCs development (1). Murine SSC originate from a founder population of Primordial Germ Cells (PGCs) formed just after embryonic day 7 (E7.0) in the extra embryonic mesoderm posterior for the primitive streak. During migration a proliferation of PGC is regulated by multiple growth factors as well as cell-cell and cell-matrix interactions. Once they arrive in the genital ridge, the PGCs are enclosed by somatic Sertoli cells and become gonocytes (2).
One of the major problems in the in vitro differentiation of germ cells is the lack of appropriate molecular markers for the characterization of the differentiated germ cells to distinguish them from the somatic cells (3). Most of the markers used for PGCs are present in Stem Cells (SCs) as well: Fragilis, Mvh, SSEA1, c-Kit (4). Therefore, it is very difficult to distinguish early germ cells from undifferentiated SCs. Meiotic and post-meiotic markers are more reliable markers, but it has been demonstrated that the progression through the meiotic process is still a challenge in the in vitro differentiation of gametes (3). The transfection of germ line cells with marked or fluorescent proteins linked to specific gene promoters (genes implicated in pluripotency or germ cell line development) enables the visualization of the cells in which the specific gene of interest is expressed during the differentiation process (5-7).
Because stem cells have unlimited potential to self-renew and produce differentiating and undifferentiating cells, heterogenous SSC transplantation offers the possibility of long-term restoration of pluripotential capability (8). Cells stimulated by growth factors, such as the remaining undifferentiated cells, showed a dramatically increase in the expression of c-Myc, and over expression of c-Myc protein is observed in a wide variety of human cancers. However, c-Myc is also expressed as part of normal cellular functions, particularly during proliferation and differentiation (9).
In this study, we introduced a novel method of in vitro SSC development. The differentiated PGC-like cells were purified by selection based on the expression of a pre-meiotic germ cells marker. The Stra-8, expressed in SSC, appeared to be a useful candidate. We used a vector with a surface reporter maker. This vector based on the expression on the cell surface of a protein that is not present in the normal mouse, does not interfere with cellular function and is efficiently recognized by monoclonal antibodies. This method facilitates identification of safely differentiated germ cells in vitro.
Materials and Methods :
Stem cell preparation, expansion, and differentiation:
Bone Marrow Stromal Cells (BMSCs) were aspirated from the tibia and femur of 5 (6-8 weeks old) male NMRI mice and were cultured according to the method described by Jamous et al. (10). Bone morphogenic protein (BMP)-4 (25 ng/ml; Chemicon, USA) was added to the fourth-passage BMSCs daily for 4 days based on the method described by Mazaheri et al. (11). PGC-like cells were enriched using a co-culture system with 3 µM retinoic acid (RA) [Sigma, Germany]. The inserts [Grainer, Germany] were placed in a 24-well plate [Grainer, Germany] containing mitomycin-treated MEF cell line (STO: C537, NCBI) and cultured for 7 days (unpublished data). CD4HAglo construct was used for purification of premeiotic differentiated cells by magnetic-activated cell sorting (MACS: Meltenyi Biotec, Germany) based on Stra-8 expression.
To induce purified Stra-8+ cells to SSCs, four groups were designed: a control (C; Dulbecco’s Modified Eagle’s Medium [DMED: Gibco, UK] supplemented 10% Fetal Bovine Serum [FBS: Gibco, South USA]), an inducer cocktail (IC; basic fibroblast growth factor [bFgF: Millipore, UK] 1 ng/ml+RA 3 µM+Leukemia Inhibitory Factor [LIF: Millipore, UK] 103 unit/ml), a co-culture system (CO; a feeder layer of Sertoli cells) and an inducer cocktail was followed by the addition of a feeder layer of Sertoli cells (IC+CO). 1×105 cells/ml were seeded into a 24 well tissue culture plate and treated with above experimental supplements during 2 days (Figure 1).
The expression of pluripotency genes (POU domain class 5 transcription factor 1 [Pou5F1], Nanog, c-Myc) and germ line genes (mouse vasa homolog [Mvh], piwi-like homolog 2 [Piwil2], stimulated by retinoic acid [Stra]-8) in purified Stra-8+ cells and four experimental groups (C, IC, CO & IC + CO) were analyzed by quantitative polymerase change reaction (qPCR).
Cell transfection using a retroviral system
Plasmid construction: The expression of a foreign neutral surface marker to purify cells that express a transgene was performed using the Stra8-CD4HAglo construct. This construct was previously described (12) and was a kind gift from Dr. Cuzin (University of Nice Sophia-Antipolis). Transfection with lipofectamine 2000 (Invitrogen, USA) was performed according to the manufacturer’s guidelines. Initially, cells were plated in 96-well plates at 5×103 cells/well and allowed to grow overnight to achieve 70-80% confluence. Cells were transfected at different DNA: lipofectamine 2000 ratios ranging from 1-3 µl lipofectamine 2000 and 0.5-1.5 µg DNA. The reagent/DNA mixture was added to OptiMem medium (Invitrogen, USA) and the cells incubated for 4-6 hr. Colonies resistant to G418 (400 mg/ml: Invitrogen, UK) were selected. Resistant colonies were analyzed by immunocytochemistry and flow cytometry to detect CD4 (Abcam, UK); colonies that contained the Stra8-CD4HAglo construct were isolated by MACS and cultured in a differentiated state. Cultures were proliferated in the above described medium for differentiation of PGC-like cells into SSC-like cells. For optimization of the transfection protocol, the expression of GFP in mBMSCs was analyzed by fluorescence microscopy using a pCMV-Myc-Cyto-GFP plasmid (Addgene, USA) as a positive control for efficiency of transfection.
Magnetic activated cell sorting (MACS): Immunomagnetic isolation of CD4+ cells from transfected cells was performed using a CD4 Positive Isolation kit (130-045-101; Miltenyi Biotec, Germany) according to the manufacturer's instructions. Purified Stra-8+ cells were analyzed by immunocytochemistry and flow cytometry to detect CD4.
Immunocytochemistry:
Purified Stra-8+ cells were fixed in 4% neutral buffered paraformaldehyde. Human T cells lymphocyte were isolated for immunocytochemical analysis as a control positive. Anti-CD4 antibody conjugated with FITC (0.1 μg/10 μl; AB86886, Chemicon, UK) was used for immunocytochemical staining. The images were captured using a Zeiss LSM 5 fluorescent microscope.
Quantitative analysis of gene expression:
Total RNA was extracted from the purified PGC-like cells (Stra-8+ cells) and SSC-like cells in four experimental groups using RNX-Plus™ (Cinnagen, Iran) according to the manufacturer’s recommendations. To eliminate genomic contamination, RNA was treated with DNase I using a kit (EN0521; Fermentas). Concentrations of RNA were determined by UV spectrophotometry (Eppendorff, Germany). The cDNAs were synthesized from 500 ng DNase-treated RNA samples with a RevertAid™ First Strand cDNA Synthesis kit (K1622; Fermentas, Germany) using oligo (dT) primers. For PCR reactions, primers were adapted from other primers (designed by the NCBI website) (6,7,13,14), and synthesized by Cinnagen (Table 1).
PCRs were performed using Master Mix and SYBR Green I in an Applied Biosystems, StepOne™ thermal cycler (Applied Biosystems, USA). The PCR program started with an initial melting cycle for 5 min at 95°C to activate the polymerase, followed by 40 cycles of melting (30 sec at 95°C), annealing (30 sec at 58°C) and extension (30 sec at 72°C). The quality of the PCR reactions was confirmed by melting curve analyses. Efficiency was determined for each gene using a standard curve (logarithmic dilution series of cDNA from the testes). For each sample, the reference gene (β2M and β-actin) and target gene were amplified in the same run. Reference genes were approximately equal. The target genes were normalized to a reference gene and expressed relative to a calibrator (purified PGC-like cells).
Statistical analyses:
Data were presented as mean±SD (standard deviation) and were analyzed using One-way repeated measure analysis of variance (ANOVA) followed by Tukey’s post hoc test. The statistics software package SPSS Version 16.0 was used to perform the calculations. If p<0.05 then the result was judged to be significant. For the real time PCR data the logarithmic values had to be converted to real values by raising 2 to the power of the ∆∆Ct value before statistical analysis was performed. Flow cytometric assays (Becton Dickenson) was done and data were analyzed with the WinMDI 2.9 software.
Results :
In vitro differentiation of bone marrow-derived PGC-like cells into SSC-like cells:
We sorted Stra-8+ cells based on the cell surface expression of CD4. When purified PGC-like cells differentiated into SSC-like cells, the expression of the Pou5F1 and Nanog were significantly decreased (p≤0.05) in the all of the experimental groups expect CO group, whereas the expression of c-Myc was significantly increased (p≤0.05) compared to purified PGC-like cells. The expression of Mvh and Piwil2 was decreased in the all of the experimental groups and that of Stra-8 was significantly increased (p≤0.05) in IC+CO group compared to purified PGC-like cells. Our results indicated that in the IC and C (without treatment or co-culture) groups, the expression of Stra-8 was significantly (p≤0.05) decreased, whereas there was no change in the CO group compared to purified PGC-like cells (Figure 2).
Our results indicated that the expression of Pou5F1 and Nanog was significantly increased (p≤0.05) in CO group, whereas there was no change compared with other experimental groups. The expression of Mvh, Piwil2 and Stra-8 were significantly increased (p≤0.05) in C, CO and IC+Co groups, whereas these genes were significantly decreased (p≤0.05) in CO and IC groups, respectively. Also, the expression of Stra-8 was down regulated in IC and C groups compared with other experimental groups (Figures 3 A and B).
In further investigations, we sorted Stra-8+ cells based on the cell surface expression of CD4. At this stage, all pluripotency genes as well as in Piwil2 were significantly decreased (p≤0.05) whereas the expression of Stra-8 was significantly increased (p≤0.05) and that of Mvh was unchanged compared to the PGC-like cells (unpublished data).
Purification of Stra-8+ cells by MACS:
We used a Stra8-CD4HALglo construct, harboring coding regions of human CD4 protein under the control of promoter regions of murine Stra-8, for transfection to induce the expression of human CD4 in PGC-like cell enrichment suspensions. We purified and cultured Stra-8+ cells using a procedure that supports expansion of isolated cells. After induction with RA (final concentration 3×106 M for 7 days with feeder cells), we identified a small population of cells (22%) within cultured adherent PGC-like cells that expressed human CD4. However, we purified approximately 80% CD4+ cells using MACS. Our observations are shown in figure 4.
Discussion :
Efficient derivation of SSCs from different sources of stem cells in vitro has been challenging in the treatment of male infertility (6,15-17). In this study, we reported a protocol of growth factor addition for differentiation followed by transfection and sorting for purification and a co-culture mimicking in vivo conditions to obtain SSC-like cells. The heterogenicity of the cells led us to sort them based on the expression of Stra8, which is regulated by RA. Adult primary cells are, in general, difficult to transfect with DNA, and BMSCs derived PGC-like cells are no exception. Importantly, for in vitro analysis of BMSCs derived PGC-like cells, the introduction of DNA has to be mediated by a technique that does not affect their proliferation and differentiation (18).
Nayernia et al showed that after RA induction (final concentration of 10−5 M), expression of Stra8-EGFP was detected in approximately 60% of all cells; they observed the expression of Stra8-EGFP in 90% of all cells after twice FACS purification (19). Furthermore, they revealed an indication of permanent activation of the Stra-8 promoter as well as differentiation into male germ cells. Our results showed that the transfection efficacy of lipofectamine 2000 after optimization was approximately 40% using a pCMV-GFP vector as a control, only 22% mBMSC-derived PGC-like cells could express the human CD4 marker. This point is important as the structure of the DNA in ESCs is very loose and could be more easily affected by inductive factors than adult mesenchymal stem cells (20). Thus, the source of stem cells used for transfection is the reason for the difference in the results obtained from our study and that from Nayernia et al. However, ESCs raise ethical issues and are more affected by transfection method than BMSCs; these problems have been solved or minimized in our study. Our cells exhibited 80% purity in the expression of Stra8 gene, which is activated by RA.
In the differentiation step we used inducer cocktail with and without Sertoli cell co-cultures which significantly increased the expression of Stra8. Sertoli cells play an essential role in germ cell development in vivo and in vitro, forming niches for germ cells which allow a certain number of germ cells to reside or repopulate the seminiferous tubules, limiting the expansion of spermatogonial population and providing essential factors for growth/proliferation and differentiation of germ cells into spermatozoa (21,22). Sertoli cells support the differentiation of germ cells into SSCs by producing BMP-4 and Activin A. Receptors of LIF and bFGF are expressed in normal mature mice SSCs (23,24). These growth factors as well as SCF could functionally differentiate PGCs into SSCs in gelatin-coated plates (7,16,17). The combination of SCF and LIF induces the proliferation and viability of PGCs (25,26). The addition of LIF, RA, and forskolin differentiated PGCs from ESCs (27), and the co-culture with Sertoli cells with LIF and RA led to the formation of germ cell colonies (28). bFGF triggers the bFGF-1 receptor, making Sertoli cells to secrete growth factors that in turn affect the PGCs, inducing expression of cell adhesion molecules such as α6 and β1 integrins to suppress migration and initiate differentiation (27). The expression of Stra-8 is significantly increased because of the effects of RA.
Our data revealed a significant increase in Piwil2 expression in Sertoli cells group compared to another groups. Piwil2 is sharply expressed in spermatids and partially in primary spermatocytes on day 14 post-fertilization and acts as a germ line–sertoli–germ line signaling loop. Piwil2 knockout mice have defective spermiogenesis initiation (29-31). The up regulation of Piwil2 occurred with Nanog & Pou5F1 in Sertoli cell group compared to another experimental differentiation groups. Synergistic Nanog and Pou5F1 control each other expression, regulating self renewal in stem cells as well as the development of germ cells (32). Nanog is expressed in PGCs until the end of migration to the genital ridge, at which point it is suppressed while differentiation is initiated (33). At the differentiation stage, the expression of Nanog was increased because of the inhibition of P53 bonding to the gene promoter (34).
Furthermore, Sertoli cells could secrete Kit-ligand (steel factor) (35), which affect the proliferation, and enrichment of PGCs after cultured in media supplemented with 10% fetal bovine serum is estimated to contain 3.6×10_8 M RA (36). These cells could release LIF, which is important for the proliferation, enrichment (in a dose-dependent manner), and self-renewal of PGCs (28). The increase in the expression of Piwil2 is attributed to its role in the regulation of self-renewal in germ cells and SSCs (29,30). On the other hand, Sertoli cells could support the stemness of PGC-like cells without differentiation to SSC-like cells compared to group of Sertoli cells following cocktail. Piwil2, Pou5F1 & Nanog were expressed during proliferation of the germ cell development. During the differentiation stage of PGC-like cells to SSC-like cells, there was a significant decrease in Piwil2, Pou5F1 & Nanog. Our results revealed that the germ line–sertoli–germ line signaling loop in the IC+Co group could support differentiation of PGC-like cells to SSC-like cell.
The expression of Mvh and Piwil2 in all of the experimental groups was lower than that in the purified PGC-like cells whereas, increased the expression levels of Stra-8 occurred toward the end of the study in IC+Co group relative to PGC-like cell. This gene is expressed in the early gonad of both male and female embryos, preventing meiosis in the germ cells when they first arrive at the gonad and come into contact with RA. Moreover, the expression of Nanog and Pou5F1 was significantly lower than that observed in Purified PGC-like cells except Sertoli cell group, but the c-Myc expression was significantly increased compared with purified PGC-like cells at the end of study in the all of experimental groups. However, a significantly decrease was observed in the expression of c-Myc after purification, while that of c-Myc increased at the differentiation step.
Conclusion :
Based on our findings, we can conclude that sorting of stem cells derived germ line cells based on promoter of specific gene could be high and efficiently safe method for cell therapy. We can eliminate undifferentiated cells with high expression of Pou5F1, Nanog and c-Myc as pluripotency genes of population cells during differentiation stage. Also, these newly differentiated cells could differentiate with LIF, bFGF, RA addition, and Sertoli cell co-culture cause increase in the expression levels of SSCs specific genes such as Stra-8.
Acknowledgement :
We are grateful to Mr Pour Beiranvand, Dr Rassoulzadegan for technical support and providing vector. We also thank Mr Langroudi for preparing the manuscript. This study was supported by Tarbiat Modares University and the Center for International Scientific Cooperation. We report no conflict of interest with regard to this study.
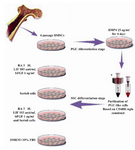
Figure 1. Schematic representation of experimental design during SSC-like cells differentiation stage from purified PGC-like cells
|

Figure 2. Real time-PCR analysis. The profile of mean cali-brated specific germ line and pluripotency genes expression (y-axis) was shown in designed different groups (x-axis) for derivation of SSC-like cells. mRNA levels were normalized with respect to β actin, chosen as an internal control. Perimordial germ cell (PGC)-like cell purified was calibrator. Differentiation stage started with PGC-like cells. Histograms show mean expression values (±SD, n=3; p<0.05). α: signifi-cant difference with other groups. Cocktile component of LIF, bFGF and RA. Sertoli was used as feeder cells
|

Figure 3. Real time-PCR analysis. The profile of mean normalized specific germ line and pluripotency genes ex-pression (y-axis) was shown in designed different groups (x-axis) for derivation of SSC-like cells. mRNA levels were normalized with respect to β actin & β2m, chosen as internal controls. Histograms show mean expression values (±SD, n=3; p<0.05). α, β: significant difference with other stages in same genes. SSC: Spermatogonial stem cells. Cocktile com-ponent of LIF, bFGF and RA. Sertoli was used as feeder cells
|

Figure 4. Schematic representation of the Stra8-CD4HLAglo fusion gene, A) RT�PCR analysis 1.4 kb promoter region of mouse Stra8 gene linked to the coding region of human CD4 in positive bacterial colony. B) Schematic representation map of vector. C) Map of CD4HLAglo construct. D) A fluorescent microscopic picture of CD4 positive cells (lymphocyte cells). E) Schematic figure of isolation of human lymphocyte cells as a CD4 positive cells. F) A fluorescent microscopic picture of CD4 positive cells after purification. G) Flow cytometric of purified CD4 positive cells
|

Table 1. Primer used for real- time PCR
α: Vasa, b; β2M, c: Stra-8 and d: Piwil2 primers were reported by Toyooka et al (2003), Boroujeni et al (2008), Nayernia et al (2004) and Lee et al (2006), respectively
|
|