Production and Purification of Streptokinase by Protected Affinity Chromatography
-
 Babashamsi, Mohammad
Ph.D., Department of Medical diagnostics & Biological products, Monoclonal antibody Research Center, Avicenna Research Institute, ACECR, Tehran, Iran, P.O. Box: 19615-1177, Tel: +98 21 22432020, Fax: +98 21 22432021, E-mail: babashams@avesina.ac.ir
Babashamsi, Mohammad
Ph.D., Department of Medical diagnostics & Biological products, Monoclonal antibody Research Center, Avicenna Research Institute, ACECR, Tehran, Iran, P.O. Box: 19615-1177, Tel: +98 21 22432020, Fax: +98 21 22432021, E-mail: babashams@avesina.ac.ir
-
Department of Medical Diagnostics and Biological Products, Monoclonal Antibody Research Center, Avicenna Research Institute, ACECR , Tehran, Iran
-
Nejadmoghaddam, Mohammad Reza
-
Department of Recombinant Technology, Nanobiotechnology Research Center, Avicenna Research Institute, ACECR , Tehran, Iran
Abstract: Streptokinase is an extracellular protein, extracted from certain strains of beta hemolytic streptococcus. It is a non-protease plasminogen activator that activates plasminogen to plasmin, the enzyme that degrades fibrin cloth through its specific lysine binding site; it is used therefore as a drug in thrombolytic therapy. The rate of bacterial growth and streptokinase production was studied in condition of excess glucose addition to culture media and its pH maintenance. The streptokinase product of the bacterial culture was preliminary extracted by salt precipitation and then purified by affinity chromatography on plasminogen substituted sepharose-4B in a condition that the plasminogen active site was protected from streptokinase-induced activation. The purity of streptokinase was confirmed by SDS-PAGE and its biological activity determined in a specific streptokinase assay. The results showed that in the fed –batch culture, the rate of streptokinase production increased over two times as compared with the batch culture while at the same time, shortening the streptokinase purification to a single step increased the yield over 95 % at the chromatography stage.
Introduction :
The clinical importance of streptokinase was first noted by Tillet and Garner (1), who discovered that this bacterial protein caused the lysis of human blood clots. It was later found that streptokinase is not an enzyme but rather a potent activator of plasminogen, the inactive precursor of plasmin (2, 3). Plasmin is the active fibrinolytic component of the circulatory system, solubilizing the fibrin network in blood clots through limited proteolysis (4, 5).
Streptokinase is currently used in clinical medicine as a therapeutic agent in the treatment of thromboembolic blockages, in-cluding coronary thrombosis (6,7). Strepto-kinase is naturally produced and secreted by various strains of hemolytic streptococci. The best studied of these is the streptokinase from Streptococcus equisimilis, from which the secretion of streptokinase into the external medium is directed by a 26 amino acid signal peptide which is cleaved during the secretion process. The mature protein has a molecular weight of about 47 kilo Dalton (kD) and was found to be composed of 415 amino acid residues (8). Karush, Iacocca, and Harris (9) and Ogburn, Harris, and Harris (10) studied the growth of a ß-hemolytic streptococcus in con-tinuous culture with pH as a limiting factor. In these experiments, pH was controlled only by addition of buffer to the medium. The yield of cells and of some extra cellular antigens was investigated. Rosenberger and Elsden (11) studied the effect of both glucose and tryptophan limitation on growth in continuous cultures of a Streptococcus faecalis strain. Their findings indicate that, to obtain max-imal cell yield per unit energy source, the energy source should be the limiting factor.
Several methods have been reported for the purification of streptokinase obtained from the culture media of various strains of streptococci. In some cases DEAE-cellulose has been used in combination with other purification procedures (12,13,14) and a highly purified product has been obtained (13).
Other chromatographic procedures have also been used for the purification of strep-tokinase by combining more than one purification step (15,16). Castellino et al (15) re-ported the use of affinity chromatography on immobilized Di-Isopropyl phosphate (DIP)-plasmin for single step purification of strep-tokinase. This method involved the conver-sion of plasminogen to plasmin by urokinase and the inhibition of plasmin protease activity by diisopropyl fluorophosphates.
Jeong et al (17) reported an affinity chro-matography using plasminogen as a ligand. Recently we have produced a fusion recom-binant streptokinase and purified it in a single step affinity chromatography using glutathi-one as the ligand (18).
In this paper, we report the results obtained from the cell growth in Todd Hewitt Broth (THB) culture media supplied with excess glucose at optimum pH and temperature; included in this report is also the rate of streptokinase production and puri-fication by affinity chromatography on acy-lated plasminogen with ?-nitro phenyl gua-nidinobenzoate (NPGB).
Materials and Methods :
The materials used in the experiment include; Streptococcus equisimilis group C, strain H46A (ATCC 12449, USA), Todd Hewitt Broth media (THB, HiMEDIA Laboratories), Trypticase Soy Agar (TSA, BBL, USA), Lysine monohydrochloride (Sigma Chemical, USA), Hexyl resorcinol (Merck, Germany), ?-nitro phenyl guanidinobenzoate (NPGB, Sigma Chemical, USA), 3-amino-n-caproic acid (EACA, Sigma Chemical, USA), Cyanogen bromide activa-ted Sepharose 4B (Sigma Chemical, USA), Chromogenic substrate S-2251 (Chromogenix laboratories, Italy), Buffer salts, acids and bases (Merck, Germany).
Extraction of streptokinase from H46A culture the bacteria were cultured in TSA at 37°C. One of the colonies was grown in 25ml of THB at 37°C. By increasing the turbidity to the level of OD=0.6 at 600 nm, it was sub-cultured in 250 ml of broth; the activity of secreted streptokinase was determined by solid and liquid colorimetric methods (19,20). It was observed that the optimum PH for cell growth and streptokinase activity was at the neutral condition (pH=7). To improve the growth condition, the pH of the culture was maintained at 7 during incubation at 37°C for 8 hours by adding sterile 4% (w/v) glucose and 5.0 N NaOH. The culture was centrifuged for 25 minutes at 10,000 g. Prior to addition of solid ammonium sulfate to a final concen-tration of 65% (w/v), the supernatant was filtered through a 0.45 µm cellulose acetate filter. After standing at 4°C overnight, the precipitate was harvested by centrifugation at 4°C for 20 minutes at 12,000 g and dissolved in 1 ml of 10 mM Tris buffer, pH=8.0, and dialyzed against repeated changes of the same buffer. Preparation of plasminogen affinity column purified plasminogen was prepared from human plasma by lysine Sepharose affinity chromatography (21). 500 ml of human plasma was centrifuged at 3000 rpm for 1 hour at 4°C to remove residual particles present in the plasma.
The supernatant then was diluted to one liter in 0.003 M EDTA and passed through a 50 ml lysine-Sepharose column at a rate of 70 ml per hour and then washed with 0.3 M sodium phosphate at pH=7.4. Upon elution with 0.3 M sodium phosphate, 0.2 M EACA, pH= 7.4, fractions of 4 ml were collected.
The material was dialyzed against 0.1 M NaHCO3, pH=8.3 and lyophilized. The purity of plasminogan was confirmed by SDS- PAGE (Figure 1). The final yield of the process was approximately 67 mg plasmino-gen per 500 ml of plasma.
Approximately 6 mg plasminogen was coupled per ml of the cyanongen bromide-activated Sepharose 4B gel. Plasminogen was then added to the resin in 0.1 M NaHCO3 (pH=8.3), and after 2 hours at room tem-perature, the resin was washed with 0.2 M glycine, (pH=8.0), and then alternately for three times with 0.1 M sodium acetate, 0.5 M NaCL, (pH=4.0), and 0.1 M NaHCO3, (pH=8.3). Purification of streptokinase to purify the streptokinase, a 12 ml bed column of plasminogen coupled to cyanogen bromide- activated sepharose 4B was equilibrated with o.ol M Tris- HCl (pH=8.0). The immobilized plasminogen was treated with 6ml of 0.5 mM NPGB in 0.01 M Tris-HCI, (pH= 8.0). The NPGB was initially dis-solved in dimethylformamide at a concen-tration of 250 mM.
The dialyzed extract of streptokinase passed through column and the column was then washed with 32 ml of buffer containing 0.01 M Tris-HCI, I M NaCL, (pH= 8.0). Elution was done with 4 ml of 8 M urea in 0.01M Tris-HCL, (pH=8.0).
Result :
The rate of streptokinase secretion incre-ased significantly in condition of excess glu-cose addition to culture media as a result of glucose metabolism and acid production.
Later by neutralizing the acidity with NaOH, the activity of streptokinase increased (Figure 2). The results of the plasminogen (plg) purification are shown in Table 1.
The streptokinase was purified with and without plasminogen acylation. In the first experiment, we treated the immobilized plasminogen with the specific inhibitor NPGB (Table 2). Two milligram (mg) pure strepto-kinase was produced, the final product had specific activity of 249850 IU/mg. In an SDS-PAGE analysis, the eluted product showed two bands of approximately 47 and 45 kD (Figure 3, lane 2).
Discussion :
Results from batch cultures indicated that strain H46A produced relatively high yields of streptokinase when the pH in the culture was controlled within the range of 7.0 to 7.1. At pH levels lower than 6.5 or higher than 7.8, streptokinase production decreased to less than 25% of that obtained at neutral pH. The comparison of this study with other studies shows that the optimum pH for cell growth and streptokinase production is 7. By adjust-ment of culture period, glucose feeding and pH maintances with concentrated NaOH the rate of product increases up to three times.
Moreover, by using suitable amount of hexyl resorcinol, the probable infection with the pathogenic streptococcus during the pro-cess was prevented.
In NPGB acylation of the immobilized plasminogen, the NPGB binds covalently to the potential substrate binding site of plasmi-nogen, thus the active site is incapable of hydrolyzing the substrate. As a result, only 5% of the salt extracted streptokinase activity was lost during the process, thus recovering 95% of the total activity (Table 2). As shown in Figure 3, the elution from the column with immobilized acylated plasminogen (lane 2) shows two bands of 47 and 45 kD that corres-ponds to the streptokinase molecule isotypes secreted by the microorganism. These kinds of streptokinases have affinity with the Glue plasminogen, which is used as a ligand in affinity chromatography.
In the experiment where the immobilized plasminogen was not acylated with NPGB (Table 2), 10% of the streptokinase activity was lost during the process, and the final recovery was of 130%. This increase in streptokinase activity is probably due to the elution of partially degraded plasminogen that forms a stable complex with streptokinase with higher specific activity. Jeong et al reported that recovery of streptokinase in their affinity chromatography on immobilized plas-minogen to be about 64%. The results of this study show that the demonstrated purification procedure for streptokinase on NPGB acy-lated immobilized plasminogen, allows the obtainment of non-degraded products with high specific activity in a single purification step. This way, the method employing acy-lated plasminogen permits one to obtain a highly purified streptokinase with high yield. The affinity gels may be reused many times without any apparent loss of binding capacity, although NPGB reaction is repeated before each use as a precaution against additional active plasminogen and plasmin being regene-rated. This acylation remains stable for less than 4 hours under the conditions described above. We suggest a further study to get an acylation with longer stability or a complete blockage of the immobilized plasminogen binding site.
Acknowledgement :
We would like to thank the Iranian Management and Programming Organization for financial support (Grant No: 31309332).
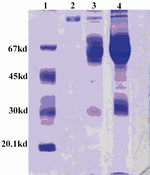
Figure 1. Evaluation of plasminogen purification by SDS-PAGE (Left to right):
1. MW marker
2. Elution dialyzed
3. Washing with 0.1 M phosphate buffer
4. Human plasma
|
![Figure 2. Variation of streptokinase [production rate] in culture media by pH regulation and glucose addition](Images/Articles/8/f2_small.PNG)
Figure 2. Variation of streptokinase [production rate] in culture media by pH regulation and glucose addition
|
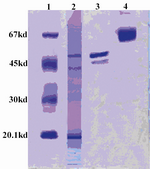
Figure 3. Evaluation of streptokinase purification by SDS-PAGE (Left to right):
1- MW marker
2- Ammonium sulfate extract
3-Purified and dialyzed streptokinase
4-BSA
|
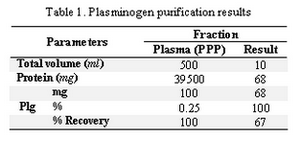
Tabel 1
|
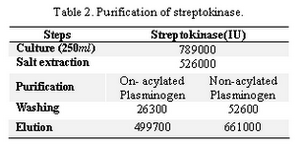
Tabel 2
|
|