In vitro and In silico Analysis of SCIN rs376349889 as a Potential Biomarker for Gastric and Colorectal Cancers
-
Vaghefinezhad, Neda
-
Department of Molecular and Cell Biology, Faculty of Basic Sciences, University of Mazandaran, Babolsar, Mazandaran, Iran
-
 Tafrihi, Majid
Department of Molecular and Cell Biology, Faculty of Basic Sciences, University of Mazandaran, Babolsar, Mazandaran, Iran, Tel: +98 1135305252; Fax: +98 1135302450; E-mail: m.tafrihi@umz.ac.ir
Tafrihi, Majid
Department of Molecular and Cell Biology, Faculty of Basic Sciences, University of Mazandaran, Babolsar, Mazandaran, Iran, Tel: +98 1135305252; Fax: +98 1135302450; E-mail: m.tafrihi@umz.ac.ir
-
Hosseinzadeh Colagar, Abasalt
-
Department of Molecular and Cell Biology, Faculty of Basic Sciences, University of Mazandaran, Babolsar, Mazandaran, Iran
Abstract: Background: Numerous research endeavors have reported altered expression of Scinderin (SCIN) in various cancer types. Single Nucleotide Polymorphisms (SNPs) represent the most prevalent form of genetic variation within the human genome which can have significant functional consequences, including cancer predisposition. Methods: This study investigated SNP-induced structural alterations in the SCIN protein and their potential effects on stability and function, using in vitro and in silico approaches. Integrating experimental and computational data provides in-sight into the role of this variant in tumorigenesis and highlights its potential as a molecular biomarker for cancer diagnosis and prognosis. Results: Out of 1,054 nonsynonymous SNPs (nsSNPs), 11 were consistently predicted to be deleterious. Among them, rs376349889 (R511G) was associated with decreased protein stability, loss of ADP-ribosylation at R511, disrupted ionic interactions, and increased hydrophobicity, all of which may impair SCIN function. Subsequently, genotyping of 200 colorectal cancer and 200 gastric cancer samples for the rs376349889 SNP was performed using High-Resolution Melting (HRM) technique in compared to a matched control group. Conclusion: The findings revealed a considerable difference in the allelic prevalence of the rs376349889 SNP between cancer patients and control samples. Notably, the GG genotype was linked to a higher susceptibility to both gastric and colorectal cancers (p<0.0001). These results suggest that rs376349889 may influence SCIN-related oncogenic mechanisms and could serve as a promising biomarker for identifying or evaluating the risk of gastrointestinal cancers at an early stage.
Introduction :
Gastric Cancer (GC) is one of the most frequently diagnosed and difficult-to-treat cancers, globally. This complex disease varies significantly from person to person, with each case often displaying unique genetic and molecular characteristics. Such individuality highlights the intricate nature of GC and the need for personalized approaches to treatment 1. Gastric cancer poses a significant health burden, worldwide, ranking as the fifth among cancers (accounting for 5.6% of cases) and the third leading cause of cancer-related deaths (7.7%) 2,3.
Colorectal Cancer (CRC) ranks third in global cancer incidence and is the second most frequent cause of cancer-related mortality 4. The overwhelming majority of CRC cases fall under the category of "sporadic," denoting the absence of any recognizable hereditary syndromes, with only a small fraction (2-8%) attributed to hereditary factors 5. Colorectal cancer is believed to develop due to a combination of inherited genes, environmental factors, and lifestyle 6.
Scinderin (SCIN), a protein named after the Latin term "scindere" meaning "to cut," functions as a Ca2+-dependent regulator of actin filament breakdown, belonging to the gelsolin superfamily 7. Its involvement extends to various physiological conditions such as exocytosis, megakaryopoiesis, autoimmune conditions, and tumorigenesis. Through the modulation of F-actin dynamics, Scinderin orchestrates the translocation of secretory vesicles and impacts cell migration by governing the process of Epithelial-Mesenchymal Transition (EMT) 8,9.
Single Nucleotide Polymorphisms (SNPs) represent one of the major forms of genetic variation within the human genome that is correlated with an individual's genetic predisposition to cancer 10-12. SNPs located in coding regions include synonymous and non-synonymous mutations that synonymous mutations have the potential to influence the expression of the protein by modulating post-transcriptional modifications, translation rates, and various additional processes. Conversely, non-synonymous SNPs (nsSNPs) induce alterations in the protein structure, as well as its physical and chemical attributes, including stability and solubility, thereby affecting its functionality 13.
Investigating genes containing SNPs contributes significantly to early cancer detection and the customization of therapies. To date, no functional investigations have explored the impact of SCIN polymorphisms on gene regulation or functionality in gastrointestinal cancer.
The present study aimed to evaluate a SNP in the SCIN gene as a potential biomarker for cancer. To this end, a combination of in vitro and in silico approaches was employed to assess the potential effects of this genetic variation on structural stability, and protein function. For this investigation, focus was made on two cancers, GC and CRC, selected due to their physiological and anatomical similarity, which facilitates comparative analysis and may reveal shared molecular mechanisms underlying tumor development. For this purpose, the HRM method was applied to evaluate the correlation between SNP rs376349889 and the susceptibility to GC and CRC. The methodology employed in the current study is briefly demonstrated figure 1. This study represents the first comprehensive investigation of the functional and structural nsSNPs within the Scinderin protein, utilizing both in silico and in vitro approaches. Nonetheless, further clinical research involving diverse ethnic groups is warranted to confirm these results.
Materials and Methods :
Scinderin protein profiling and retrieval of SNP data
To gather information on the SCIN gene (UniProt ID: Q9Y6U3), including the amino acid sequence and functional attributes, we accessed the UniProtKB database (http://www.uniprot.org/uniprot/). For a deeper understanding of the Scinderin protein, InterPro database (https://www.ebi.ac.uk/interpro/), was utilized which helps to identify specific domains, motifs, and other conserved features. This allowed the authors to explore the protein's structure and potential functions by referencing a comprehensive collection of protein families and functional site data 14.
Additionally, Scinderin structural property prediction was performed using RaptorX property, a web server that predicts structural properties of protein sequences without relying on a template (http://raptorx6. uchicago.edu/StructurePropertyPred/predict/) 15.
The data regarding SNPs of the human SCIN gene were acquired from the dbSNP-NCBI database (http:// www.ncbi.nlm.nih.gov/SNP/). Subsequently, the ns-SNPs were excluded to facilitate further investigations 16.
Identification of potentially deleterious SNP variants
To assess the potential impact of SNPs in the SCIN gene's coding sequence, multiple bioinformatic platforms—SIFT, PANTHER, PolyPhen-2, SNAP2, PredictSNP, and Align GVGD were employed.
Analyzing potential deleterious effects of nsSNPs was conducted using the Sorting Intolerant from Tolerant (SIFT) web-based tool (http://sift.jcvi.org/). SIFT evaluates the impact of each SNP by considering both sequence similarity and the physical properties of the protein. The SIFT scores ranged from 0 to 1, where a score lower than 0.05 suggests the adverse influence of nsSNPs on protein structure or function 17.
The PolyPhen-2 tool (http://genetics.bwh.harvard. edu/pph2) evaluates how changes in amino acids influence protein structure and functions through sequence-based characterization. Predictions were given as probability values divided into three distinct groups: "benign", "possibly damaging", and "probably damaging" with a cutoff value that has been set for probably damaging, with a score exceeding 0.95 18.
The PANTHER-PSEP web server (http://pantherdb. org/tools/csnpScoreForm.jsp) categorizes the impact of nsSNPs on protein activity by considering evolutionary relationships, molecular functions, and protein-protein interactions. It examines amino acid changes through evolutionary conservation scores obtained from aligned sequences of diverse, related proteins 19.
The SNAP2 web server (https://www.rostlab.org/ services/SNAP/) relies on neural network algorithms to make predictions regarding the influence of nsSNPs on secondary structure of proteins. Additionally, a solvent accessibility comparison is performed between the native and altered protein structures to help separate functionally relevant changes from neutral ones 20.
The Align GVGD online tool (http://agvgd.hci.utah. edu/) is grounded in biophysical characterization. By integrating multiple sequence alignments of proteins with amino acid biophysical characteristics, this tool effectively forecasts the impact of a missense change as either benign or harmful. These mutations are classified into seven groups: C0, considered the most probable to be neutral or benign, C15, C25, C35, C45, C55, and C65, identified as the most likely to be deleterious or pathogenic 21.
The PredictSNP online tool (https://loschmidt. chemi.muni.cz/predictsnp1/) compiles data inputs from multiple sources to determine the potential impact of amino acid changes. By adopting this collaborative approach, PredictSNP offers a more comprehensive view and improves predictive accuracy compared to individual predictors 22.
Different tools use various algorithms, so single nucleotide polymorphisms identified by all of them as having harmful effects-such as changes in structural stability, energy levels, and surface availability-were selected for further analysis.
Assessment of structural stability alterations in mutant proteins
Protein function is linked to its stability, emphasizing the importance of identifying alterations in protein stability caused by nsSNPs. Five computational tools—IMutant2.0, MutPred2, MUpro, NetSurfP-2.0, and HOPE—were employed to evaluate the stability of the mutated protein.
IMutant2.0 (http://folding.biofold.org/i-mutant/im-utant2.0.html) is an SVM-based tool used to assess whether a modification in an amino acid affects protein stability. The analysis of nsSNPs using this tool was conducted under specific conditions: a temperature of 25°C and a pH of 7.0, while relying on the ΔΔG value and binary classification system for the prediction process. The assessment relies on ΔΔG values, reflecting the difference in Gibbs free energy between the wild-type and mutated forms. Additionally, an RI value, denoting the reliability index, was assigned to each result generated by the tool. A ΔΔG value below 0 indicates reduced protein stability, whereas a value above 0 signifies increased stability 23.
MUpro (http://mupro.proteomics.ics.uci.edu/) is a web-based tool is utilized to predict protein stability alterations caused by SNPs, employing Support Vector Machine (SVM) and neural networks algorithms. Stability predictions can be made using this tool without the need for tertiary structure input. The results indicate the energy variation due to an amino acid substitution, presented as a ΔΔG value with a confidence rating from -1 to 1. A ΔΔG value below 0, signifies reduced protein stability, while a value above 0 indicates increased stability 24.
To evaluate the predicted structural and functional consequences of amino acid substitutions, the MutPred2 server (http://mutpred.mutdb.org/) was utilized. Amino acid modifications can affect protein functionality in various ways, including structural destabilization, disruption of macromolecular binding, and loss of Post-Translational Modifications (PTM) sites. These alterations in molecular processes often play a significant role in modifying the phenotypic effects of a protein. Using the MutPred2 algorithm, SNP pathogenicity levels are assessed, and a wide range of structural and functional modifications resulting from these amino acid changes are predicted. Additionally, empirical p-values are provided for each modification 25.
To predict the secondary structure and exposure level of amino acids on the protein surface, the NetSurfP server (http://www.cbs.dtu.dk/services/NetSur-fP/) was employed. Using a neural network-based architecture that integrates convolutional and long short-term memory networks, this tool analyzes the amino acid sequence to predict exposure level of amino acids on the protein surface, secondary structure, and structural changes at each position. The Relative Surface Accessibility (RSA) threshold is set at 25%; residues with RSA >25% are classified as exposed 26.
The HOPE server (https://www3.cmbi.umcn.nl/ hope/) was employed to assess the impact of deleterious SNPs on the three-dimensional protein structure. HOPE provides insights into how SNPs affect properties such as residue size, electrical charge, hydrophobicity, spatial conformation, bond alterations, and functional characteristics, with integrated 3D visualization. Additionally, HOPE evaluates the conservation score of the altered residue using HSSP multiple-sequence alignment 27.
Protein modelling and structural analysis
Both the wild-type and mutant proteins, which exhibited a significant impact following a point mutation based on initial analysis, were modeled using the trRosetta algorithm. This online tool predicts the structural arrangement of proteins (https://yanglab.nankai.edu.cn/ trRosetta/) through a deep learning-based ab initio folding approach 28.
The mutated models were evaluated using the TM-align tool (https://zhanglab.dcmb.med.umich.edu/TM-align/) to assess structural deviations between the native and mutant models. TM scores and Root Mean Square Deviation (RMSD) values were computed by superimposing the protein structures to identify structural similarities. TM scores range from 0 to 1, with a value of 1 signifies a a perfect structural match. A TM score between 0.0 and 0.30 suggests random structural similarity, whereas a score of 0.5 or higher indicates that the structures share the same fold 29.
Computational analysis of nsSNP-induced changes in protein stability and interactions
The DynaMut server (http://biosig.unimelb.edu.au/ dynamut/) was employed to predict how nsSNPs influence protein stability and molecular interactions. This server predicts structural changes by analyzing protein flexibility and dynamics. DynaMut utilizes Normal Mode Analysis (NMA) to calculate the difference in Gibbs free energy (ΔΔG) between the Wild-Type (WT) and Mutant (MT) protein structures. It also evaluates the change in flexibility ENCoM-based vibrational entropy differences (ΔΔSVib). DynaMut further provides insights into how mutations affect protein conformation, flexibility, and stability, along with visualizations of dynamic behavior 30.
Gene-gene interaction analysis
The potential associations among gene networks and gene interactions were predicted using the GeneMANIA online tool (http://genemania.org/). This information includes gene-gene and protein-protein interactions, structural resemblance of protein domains, co-expression, pathways, and colocalization. When given a list of query genes, GeneMANIA expands this list by incorporating functionally similar genes based on available genomics and proteomics data. The gene of interest was analyzed using GeneMANIA to visualize and explain the interconnections among the identified genes 31.
Protein-protein interaction (PPI) network analysis
Amino acid sequence alteration can lead to conformational changes in proteins, potentially affecting their functional properties. As a result, the mutated protein may interact with different partners, resulting in observable phenotypic effects. To explore the interaction patterns of SCIN with various proteins, the STRING server (http://string-db.org/) was utilized. This platform offers a comprehensive analysis of protein-protein interactions, incorporating both predicted and experimentally validated interactions 32.
Sample preparation
The study population comprised 200 GC patients, 200 CRC patients, and 200 healthy controls. Blood samples were collected from all participants—following informed consent at Sayed-Al-Shohada and Al-Zahra hospitals in Isfahan, Iran. Approximately 5 ml of peripheral blood was drawn from all enrolled individuals and collected in tubes containing EDTA as an anticoagulant. Genomic DNA was isolated using the GeneAll Exgene kit (Korea) in accordance with the manufacturer’s protocol.
Real-time PCR and high-resolution melting (HRM) analysis
The sequence of SNP rs376349889 was identified using the GeneBank database. The sequence and additional information related to this SNP were obtained from NCBI. The online tool Primer3Plus (https://www. bioinformatics.nl/cgibin/primer3plus/primer3plus.cgi) was used to design a specific pair of primers. Primer melting temperatures (Tm) and design suitability parameters were assessed using OligoAnalyzer, and primer specificity was assessed using the Primer-BLAST online tool (https://www.ncbi.nlm.nih.gov/tools/ primerblast/).
The HRM technique was employed for amplification reactions using a MIC qPCR cycler (BioMolecular Systems, USA). Each reaction was prepared in a final volume of 10 μl containing 2 μl of cDNA (25 ng/μl), 2 μl of Evagreen, 0.5 μl of forward primer (5'-CAAAGA-AAGGAGGTCAGGCAC-3', 5 μM), and 0.5 μl of reverse primer (5'-TGGTGATAGATGCCAGGT TTCT-3', 5 μM). To validate the PCR results, each genotype was sequenced using the HRM technique 33.
qRT-PCR was performed for 45 cycles with the following temperature conditions: 15 min of preincubation at 95°C, 15 s of denaturation at 95°C, 20 s of annealing at 60°C, 20 s of extension at 72°C, followed by a melting curve analysis conducted from 60°C to 95°C. HRM analysis was performed using the data derived from the melting curves by the Mic’s qPCR analysis software 34.
Statistical analysis
Data analysis was performed using GraphPad Prism software (version 9.5.1, GraphPad software, San Diego, CA). Odds Ratios (ORs) with 95% Confidence intervals (Cis) were calculated to evaluate risk, and a p-value <0.05 was considered statistically significant. Group differences were assessed using the chi-square test.
Results :
Scinderin protein profiling and retrieval of SNP data
The SCIN gene in humans consists of 89,463 base pairs, with its corresponding protein composed of 715 amino acids. An investigation into the presence of distinct functional domains within the SCIN protein_such as gelsolin_S1_like (10-121), VILL_6 (135-227), gelsolin_S3_like (246-346), gelsolin_S4_like (390-486), gelsolin_S5_like (508-598), and gelsolin_S6_like (613-711)-was conducted using resources from the InterPro and UniProtKB databases.
The structural data derived from the UniProtKB archive revealed specific domains that encompassed an actin-severing segment (1-363) and a Ca2+-dependent actin-binding region (364-715).
The RaptorX server predicted 18% α-helix, 28% β-sheet, and 53% coil for the SCIN protein. Additionally, three categories of residue-specific solvent accessibility were identified: exposed (40%), medium (33%), and buried (26%), Furthermore, 38 residues (5%) were predicted to be disordered. A total of 38,785 SNPs associated with the SCIN protein were retrieved from the dbSNP database, considering only nonredundant entries. The identified SNPs were assigned to distinct functional categories, including in-frame deletion 8, in-frame indel 2, in-frame insertion (3), start codon variant 7, intron (33343), missense (1054), noncoding transcript variant (6059), and synonymous (417) SNPs. The majority of SNPs were found within intronic regions, subsequently by noncoding transcript variants and nsSNPs. The distribution of these SNPs is shown in figure 2.
Identification of potentially deleterious SNP variants
To predict the pathogenicity or deleterious effects of nsSNPs, five bioinformatics tools—PolyPhen-2, SNA-P2, Align GVGD, SIFT, and PANTHER—were utilized, each applied according to its respective predefined criteria. Consequently, 11 nsSNPs out of 1,054 were unanimously predicted to be deleterious by all five tools. When analyzed by PredictSNP, 9 mutations were classified as severely deleterious (Table 1).
Assessment of structural stability alterations in mutant proteins
MUpro and I-Mutant2.0 assessed the influence of various nsSNPs on the stability of the SCIN protein. The results indicated a general decrease in stability across all nsSNPs, except for the P382L SNP, which was predicted to enhance stability based on analysis conducted using IMutant2.0 (Table 2).
Prediction of structural effect of deleterious SNPs
Predictions generated by MutPred suggested potential gains of acetylation, modifications in metal binding, acquisition of catalytic sites, loss of ADP-ribosylation, and potential gains of phosphorylation. These predictions were accompanied by p-values and probability scores. A MutPred2 score, derived from machine-learning neural network algorithms, was also reported. This score estimates the likelihood of structural and functional changes, including intrinsic disorder, disruptions in ordered interfaces, metal binding, and helix modification. The score varies between 0 and 1, with a higher value indicating a greater probability that SNP-induced changes contribute to disease-related molecular mechanisms.
As a result, all selected nsSNPs were considered highly plausible hypotheses, except for the T131R, R271T, and P382L mutations, which were classified as confident hypotheses (Table 3).
Computational analysis of nsSNP-induced changes in protein stability and interactions
The solvent accessibility of the SCIN protein was evaluated using the NetSurfP online tool. Notable transitions between exposed and buried states were observed. The results revealed that the M384T variant caused a transition from a buried to an exposed state, while the P382L variant resulted in a shift from an exposed to a buried state. Additionally, other mutations resulted in alterations in Relative Solvent Accessibility (RSA) and Absolute Solvent Accessibility (ASA) values, suggesting changes in solvent exposure (Table 4).
The mutations were further analyzed using the HOPE tool. The results showed that, among the seven mutations with a MetaRNN score exceeding 0.5, the score of five reflected modifications in the size of the amino acid, four indicated an alteration in charge, and six affected hydrophobicity. Additionally, various other biochemical properties were influenced by amino acid substitutions. According to the HOPE prediction report, the rs376349889 variant, which involves a glycine substitution (R511G), was particularly noteworthy. This substitution introduced flexibility due to glycine's structural properties, potentially disrupting the necessary rigidity at that specific position within the protein. Moreover, compared to the wild-type residue, the mutant residue was reduced in size. The mutant residue was neutral and showed increased hydrophobicity compared to the positively charged wild-type residue. Furthermore, the wild-type residue formed a salt bridge with residue number 527 (glutamic acid), which could be disturbed by the charge difference introduced by the mutant residue. The site of the mutation falls within a segment of residues identified in UniProt as a specialized region responsible for Ca2+-dependent actin binding. Alterations in amino acid properties could potentially interfere with this region and impact its functionality (Table 5).
Structural analyses of protein mutations
Due to the absence of a full-length SCIN protein 3D structure in the Protein Data Bank (PDB), the FASTA-formatted amino acid sequence, along with mutated variants, was uploaded to the trRosetta server to generate 3D models of the SCIN protein (Figure 3A). Similarly, the same procedure was applied to generate a model for the mutated protein (Figure 3B). The predicted wild-type and mutant protein models had Template Modeling (TM) scores of 0.710 and 0.706, respectively. A TM-score greated than 0.5 indicates considerable similarity in three-dimensional conformation between the predicted protein models and their template structures. Both models demonstrated TM-scores above 0.5, suggesting reliable structural predictions.
The degree of structural similarity between the native and mutant models was assessed using TM-score and RMSD values via the TM-align tool (Figure 4). The predicted mutant model obtained a TM score of 0.48, suggesting notable structural divergence from the wild-type protein. Additionally, RMSD analysis was conducted to quantify structural deviations in the mutant model relative to the wild-type protein, revealing a value of 6.24, indicating considerable structural instability associated with this high-risk nsSNP.
Prediction of mutation-induced structural and stability changes using DynaMut
The impact of the mutation on protein stability and dynamics was assessed using the DynaMut web server. The ΔΔG values obtained from different prediction algorithms indicated a generally destabilizing effect. Specifically, the overall DynaMut ΔΔG was –0.068 kcal/mol, while ENCoM (–0.080 kcal/mol), mCSM (–1.451 kcal/mol), and DUET (–1.116 kcal/mol) consistently predicted destabilization. In contrast, the SDM method suggested a slight stabilizing effect with a ΔΔG of +0.370 kcal/mol. Additionally, the change in vibrational entropy energy (ΔΔSVib ENCoM=+0.100 kcal/mol·K) revealed an increase in molecular flexibility in the mutant structure compared to the wild type. To further investigate the destabilizing effects of the mutation, an interatomic interaction analysis was conducted. To further investigate the destabilizing effects of the mutation, an interatomic interaction analysis was conducted. The wild-type residue (green) formed multiple stabilizing interactions, including ionic interactions and hydrogen bonds, whereas the mutant residue showed reduced bonding interactions, indicating decreased local stability (Figure 5).
Analysis of gene-gene and protein-protein interactions
The GeneMANIA tool was utilized to evaluate gene-gene interactions involving the SCIN protein, revealing an association with the TMOD2 gene. Additionally, figure 6 illustrates the co-expressed genes and their roles in achieving similar functions or sharing common protein domains. The investigation revealed interactions between the SCIN protein and several proteins involved in cytoskeletal functions, including HCLS1 (Hematopoietic Lineage Cell-Specific Lyn Substrate 1), CTTN (Src substrate cortactin), TLN1 (Talin-1), WASL (Neural Wiskott-Aldrich syndrome), and TLN2 (Talin-2). Additional protein interactions are depicted in figure 7.
Analysis of HRM results
We examined the rs376349889 (C>G) variant in both gastric and colorectal cancer samples, utilizing genotypic data obtained from the HRM test. Genotyping was conducted on 200 patients diagnosed with CRC and 200 patients diagnosed with GC (both female and male), focusing on the rs376349889 variant. The allele frequencies and genotype distributions for each group are summarized in table 6. A marked difference in rs376349889 frequency was observed among GC patients, CRC patients, and the control group. The primary element that differentiates wild type (CC), homozygous (GG), and heterozygous (GC) genotypes is the variation in melting temperature and TM point, as illustrated in figure 8. Analysis revealed that the G allele was more prevalent in GC samples compared to the wild-type C allele. Additionally, the GG genotype was more common in gastric cancer samples than the CC genotype when compared to healthy control samples. Comparable patterns were also detected in CRC samples (Table 6).
Discussion :
Global cancer statistics from 2022 indicate that approximately 20 million new cancer cases were diagnosed, resulting in nearly 10 million deaths attributed to the disease. Projections for 2050 forecast an annual incidence of 35 million new cancer cases, reflecting a 77% increase from 2022 figures 35. Notably, gastrointestinal cancers, including gastric and CRC, are significant contributors to cancer-related mortality 36.
Scinderin, an actin-binding and actin-severing protein, is essential for the regulation of cytoskeletal dynamics 7. Several studies have reported altered SCIN expression in various cancer types, suggesting its potential involvement in tumorigenesis, acting as either a tumor suppressor or an oncogene depending on the tissue. Current evidence is limited and sometimes conflicting, with studies reporting both increased and decreased expression associated with malignancy 37-39. In megakaryoblastic leukemia, ectopic expression of SCIN reorganizes actin filaments via Rho GTPases (CDC42, Rac1), leading to differentiation, maturation, and apoptosis with release of platelet-like particles. These effects are accompanied by suppression of proliferation and tumorigenesis, partly through modulation of PI3K/AKT and MAPK/ERK signaling pathways 40. These findings underscore the complexity of SCIN’s function in tumorigenesis. Further research is needed to clarify its molecular mechanisms and evaluate its potential as a context-specific prognostic marker or therapeutic target.
In this study, nsSNPs were analyzed using six computational tools: SIFT, PANTHER, PolyPhen-2, SNAP2, PredictSNP, and Align-GVGD. This approach aimed to leverage their diverse predictive capabilities and improve filtration accuracy. SIFT classifies substitutions based on evolutionary conservation, with scores ≤0.05 considered deleterious 17. PolyPhen-2 integrates evolutionary, structural, and physicochemical features, where variants labeled probably damaging are more likely to impair protein function 18. PANTHER uses evolutionary conservation, with subPSEC scores ≤−3 indicating a higher likelihood of deleterious impact 19. SNAP2 employs neural network-based integration of sequence, structural, and physicochemical features, where positive scores suggest deleterious effects and negative scores indicate tolerated variants 20. Align-GVGD combines evolutionary conservation and amino acid chemical differences, classifying variants from C0 to C65, with higher classes reflecting greater likelihood of deleterious impact 21. PredictSNP provides a consensus prediction by integrating multiple tools, where variants classified as deleterious with high confidence are more likely to disrupt protein function 22, Due to the unique parameters of each algorithm, nine nsSNPs were consistently classified as highly deleterious across all tools, warranting further investigation (Table 1).
Protein stability is essential for maintaining proper molecular function. Missense mutations can impair a polypeptide’s ability to adopt the correct functional conformation, leading to decreased stability. Consequently, mutant proteins are more susceptible to misfolding, aggregation, or degradation. Reduced stability and misfolding are significant outcomes of damaging missense variants 41.
To assess the impact of nsSNPs on protein stability, we utilized the MUpro and I-Mutant3.0 online tools for both wild-type and mutant SCIN proteins (Table 2). These tools estimate whether amino acid substitutions increase or decrease stability, reporting complementary outputs: MUpro provides scores ranging from -1 to +1, while I-Mutant 3.0 calculates ΔΔG values (kcal/mol) along with a reliability index (RI) 23,24. The thermodynamic stability of a protein is indicated by the Gibbs free energy of folding (ΔG), which represents the energy difference between its folded and unfolded forms. When ΔΔG is positive, it implies greater stability; conversely, a negative ΔΔG indicates reduced stability 42. Our analysis showed that most nsSNPs within the SCIN gene decreased protein stability, except for the P382L variant, which was predicted to enhance stability by I-Mutant2.0.
MutPred2 was employed to evaluate the functional consequences of amino acid substitutions based on physicochemical properties such as ubiquitylation, methylation, acetylation, and changes in ordered/ disordered interfaces, generating a general deleteriousness score from 0 to 1, where scores >0.5 indicate a higher likelihood of pathogenicity 25. Six nsSNPs were predicted to induce significant structural and functional alterations, each scoring above 0.75 on the severity scale (Table 3).
Beyond functional predictions, assessing structural properties such as solvent accessibility, secondary structure, and structural disorder provides additional insights into the potential impacts of these variants. Using NetSurfP-2.0, provided insights into the secondary structure (H: helix, E: strand, C: coil), RSA, 0–1 and ASA, disorder probability (0–1, with values near 1 indicating disordered regions), and backbone dihedral angles (φ, ψ) of each amino acid, enabling detailed structural analysis 26. Among the 11 highly deleterious nsSNPs, the M384T mutation was identified as surface-exposed, while P382L was buried. Four mutations (T131R, R145L, and R146G) were located on the protein surface, potentially affecting the physiological properties of the protein, such as stability, charge distribution, and accessible surface area. Conversely, five buried nsSNPs (K306S, W440R, I492S, R511G, and S620Y) may disrupt protein folding and function (Table 4).
Structural assessment using the HOPE server revealed hydrophobicity, charge, and size variations between the wild-type and mutant amino acids, potential effects on protein structure and stability, visualizations and 3D models when structural data are available, and a summary of the likely impact on protein function 27. Seven substitutions—T131R, P271S, R306S, M384T, W440R, R511G, and S620Y—had a MetaRNN score of ≥0.5, indicating potential pathogenicity. Notably, W440R and R511G had scores exceeding 0.9, suggesting a high likelihood of functional disruption. For instance, rs376349889 results in the substitution of arginine (R) with glycine (G) at position 511. Glycine, being smaller and uncharged, differs significantly from the positively charged wild-type arginine, affecting local hydrophobicity and disrupting hydrogen bonding. In addition, a salt bridge is formed between arginine and glutamic acid at position 527—an interaction lost in the mutant protein—which may affect the actin-binding capacity of the SCIN protein (Table 5).
While residue-level analyses highlight local disruptions, investigating the impact on overall protein architecture required structural modeling. Due to the lack of a comprehensive structural model for the human SCIN protein (715 amino acids) in the Protein Data Bank, computational modeling was performed using trRosetta, which provided outputs including pairwise distance maps between all residues, predicted backbone dihedral angles (φ, ψ, ω), 3D protein models in PDB format, and quality scores indicating the confidence of the structural predictions 28. The wild-type and R511G mutant models had TM-scores of 0.710 and 0.706, respectively, as calculated by trRosetta —both values above 0.5, indicating reliable topological similarity (Figure 3) TM-align compared 3D protein structures using TM-scores, RMSD, and residue-level alignments to assess changes induced by nsSNPs, revealed a significant conformational shift in the R511G variant, evidenced by an RMSD value of 6.24 and a decreased TM score of 0.484 (Figure 4). High RMSD values (typically >2–3 Å) indicate low structural similarity, suggesting significant conformational differences that may affect protein function. In contrast, TM-scores values below the 0.5 threshold, the topological similarity is considered unreliable, suggesting that the mutation may alter the overall fold or conformation of the protein and thereby compromise its function 29.
The computational analysis by DynaMut indicated that the mutation leads to modest but consistent destabilization of the protein structure, as supported by mCSM and DUET, while SDM predictions diverged due to its reliance on sequence-based rather than structural information. Although ΔΔG values from DynaMut and ENCoM (–0.068 and –0.080 kcal/mol) were close to the neutral threshold (–0.5 to +0.5 kcal/mol) 43, the more negative values from mCSM (–1.451 kcal/ mol) and DUET (–1.116 kcal/mol) support a destabilizing effect. The positive ΔΔSVib further suggests increased molecular flexibility and reduced conformational stability. Such changes in dynamic behavior may weaken intramolecular interactions, thereby affecting the protein’s overall structural integrity and potentially altering its functional properties. Structural comparison revealed that, unlike the wild-type residue which formed stabilizing hydrogen and ionic bonds, the mutant residue showed a loss of these interactions and aromatic contacts, indicating weakened local stability and possible structural rearrangements that may compromise protein function. Gene interaction analysis revealed a strong association between the SCIN gene and various members of the GSN gene family. Prior research efforts have revealed that TMOD2 is considerably overexpressed in cells isolated from breast and prostate carcinomas 44,45. In colorectal cancer, VAV3 has been linked to increased tumor invasion and metastasis 46, while AVIL overexpression in glioblastoma stimulates cell proliferation and migration 47. Additionally, SVIL is implicated in gastric cancer progression but is downregulated in bladder cancer 48. These findings suggest that deleterious SNPs within the SCIN gene could affect broader gene networks (Figure 6). Network analysis using GeneMANIA further highlighted potential functional interactions between SCIN and these cancer-associated genes, providing insight into how deleterious nsSNPs might perturb key oncogenic and tumor-suppressive pathways.
Beyond SCIN, the analyzed signaling network includes both oncogenes and tumor suppressors. PIP5K1A, CTTN and TLN1 act as oncogenes, promoting tumor progression and metastasis 49-51. Conversely, PIP4K2A, TLN2, WASL, and HCLS1 function as tumor suppressors, regulating PI3K and Wnt/β-catenin signaling, cytoskeletal dynamics, or anti-tumor immunity 52-55. PIKFYVE additionally modulates tumor immunity by enhancing MHC class I surface expression, offering a potential therapeutic target 56. Although the roles of PIP5K1B, PIP5K1C, and TLN1 are less defined, integrating these proteins with SCIN variants highlights the complex network influencing cancer progression and may guide future therapeutic strategies (Figure 7).
Having identified SCIN's potential interaction network, we further investigated the clinical significance of specific SCIN variants. Genotypic analysis of rs376349889 (C/G) in gastric and colorectal cancer samples revealed significant associations between the GG genotype and increased disease risk. OR calculations indicated that allele G increases the likelihood of cancer by 2.825 times (GC) and 1.924 times (CRC), further supporting its pathogenic potential (Table 6).
Although SCIN has emerging roles in cytoskeletal regulation and tumor-related processes, there is still very little population-based genetic evidence directly linking SCIN variants to human disease. Thus far, the only published document comes from a melanoma epistasis study, where an intronic SCIN polymorphism (rs7798406) showed a significant interaction with a CDC42 variant (rs3117048) in influencing Breslow thickness 57. Apart from this finding, genome-wide association studies have not identified SCIN as a locus of major effect, highlighting the need for more comprehensive genetic and functional studies to better define the role of SCIN variants in cancer development and progression. Given the scarcity of current data, it is still uncertain whether SCIN polymorphisms independently affect cancer risk and prognosis, or whether their influence is conditional and mediated through interactions with other signaling networks.
Conclusion :
This research highlights the impact of single nucleotide polymorphisms in the SCIN gene, notably R511G, on protein stability and their possible involvement in cancer progression. These substitutions may disrupt the cell's internal structure, potentially promoting tumor growth in gastrointestinal cancers. While present findings strongly suggest a connection, experimental validation through laboratory studies is necessary to validate these effects. Additionally, further research should investigate how these substitutions influence the behavior and function of cancer cells. Large-scale genetic studies could also help determine whether SCIN substitutions are reliable biomarkers, facilitating earlier detection and more targeted treatments for individuals at risk.
Acknowledgement :
We would like to thank the University of Mazandaran’s Research Department.
Conflict of Interest :
The authors declare no conflict of interest.
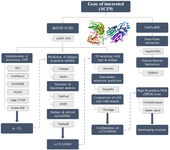
Figure 1. Flowchart outlining the steps taken to identify the rs376349889 SNP and the associated experimental designs. Integration of In vitro and In silico analyses to identify functionally significant SNPs, evaluate their effects on protein structure and stability, analyze their roles in gene-gene and protein-protein interaction networks, and ultimately identify potential molecular biomarkers for disease diagnosis, prognosis, or therapeutic applications.
|
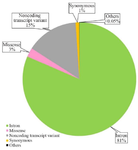
Figure 2. A graphical representation in the form of a pie chart illustrating the distribution of Single Nucleotide Polymorphisms (SNPs) within the SCIN gene, as sourced from the dbSNP database.
|
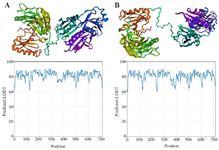
Figure 3. 3D Protein Modeling and Quality assessment of SCIN proteins using trRosetta. A) Wild-type SCIN, B) R511G mutant. Comparison of predicted local confidence (pLDDT) scores for the wild-type (A) and mutant (B) proteins, showing overall similar structural reliability with minor local variations.
|
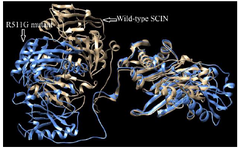
Figure 4. Superimposed structure of Wild-type SCIN and its R511G mutant generated by the TM-align tool. High RMSD (6.24) and low TM score values (0.48) indicate structural dissimilarity due to R511G mutation. The Wild-type structure is shown in gold, while the R511G mutant is shown in blue.
|
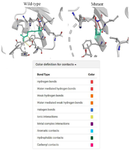
Figure 5. Comparative structural analysis of the mutant (R511G) SCIN protein in comparison to the wild-type structure generated by DynaMut. In the wild-type conformation, the residue of interest (green) establishes multiple stabilizing contacts, including ionic interactions, aromatic contacts and hydrogen bonds, whereas the mutant conformation exhibits a marked reduction in these interactions, suggesting decreased local stability and potential structural rearrangements.
|
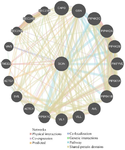
Figure 6. GeneMANIA-based analysis of SCIN SCIN gene interactions.
|
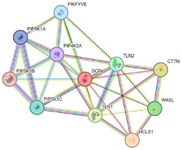
Figure 7. SCIN protein interaction network constructed via STRING.
|

Figure 8. The distinction in melting profiles among wild type (CC), homozygous (GG), and heterozygous (GC) genotypes; (A) gastric cancer patients, (B) colorectal cancer patients.
|
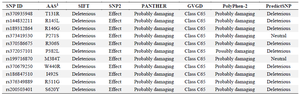
Table 1. Identification of deleterious Single Nucleotide Polymorphisms in the SCIN protein using various online prediction tools
AAS1: Amino Acid Substitution
|
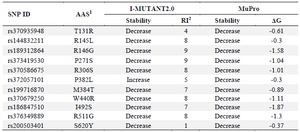
Table 2. Predictions of protein stability changes using I-Mutant 2.0 and MUpro.
AAS1: Amino Acid Substitution, RI2: Reliability Index
|
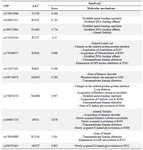
Table 3. Prediction of the functional and structural alterations using the MutPred2 server.
AAS1: Amino Acid Substitution
|
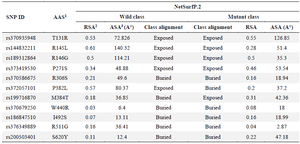
Table 4. Structural evaluations of deleterious SCIN protein variants using the NetSurfP.2 web server
AAS1: Amino Acid Substitution, RSA2: Relative Solvent Accessibility, ASA3: Absolute Solvent Accessibility
|
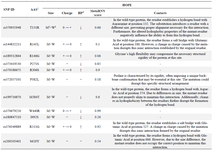
Table 5. Analysis of structural impacts of the selected mutations using the Project HOPE web server
AAS1: Amino Acid Substitution, M2: Mutant, W3: Wild, HP4: Hydrophobicity.
|
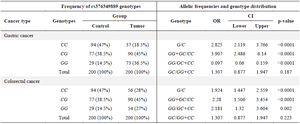
Table 6. Frequency of rs376349889 genotypes, allelic frequencies and genotype distribution in Gastric Cancer (GC) and Colorectal Cancer (CRC) patients compared with control subjects
|
|