Designing a Novel Immunotoxin against Prostate Cancer based on PE40 Toxin: An In silico Approach
-
Azdoo , Sadaf
-
Cellular and Molecular Research Center, Yasuj University of Medical Sciences, Yasuj, Iran
-
Taheri-Anganeh, Mortaza
-
Cellular and Molecular Research Center, Cellular and Molecular Medicine Research Institute, Urmia University of Medical Sciences, Urmia, Iran
-
Ahmadi, Khadijeh
-
Department of Medical Biotechnology, School of Paramedicine, Bushehr University of Medical Sciences, Bushehr, Iran
-
 Movahedpour, Ahmad
Cellular and Molecular Research Center, Yasuj University of Medical Sciences, Yasuj, Iran, Tel: +98 74 33346078; E-mail: ahmad.movahed14@gmail.com
Movahedpour, Ahmad
Cellular and Molecular Research Center, Yasuj University of Medical Sciences, Yasuj, Iran, Tel: +98 74 33346078; E-mail: ahmad.movahed14@gmail.com
-
Jamshidi , Ali
Behbahan Faculty of Medical Sciences, Behbahan, Iran, Tel: +98 74 33346078; E-mail: dani1364@gmail.com
Abstract: Background: Prostate cancer, is the second most prevalent malignant tumor and fifth leading cause of cancer-related death among men worldwide. Patients suffer from adverse side effects and low efficacy of traditional therapeutic approaches. At present, cancer-targeted therapy is a fascinating strategy of cancer therapy re-search via employing immunotoxins, which is a fusion of a targeting molecule and a killer toxin that can recognize a specific antigen on cancerous cells and trigger cell death. Methods: This study used a prostate-specific scFv and a truncated version of Pseudomonas-exotoxin to design a novel immunotoxin. After the construct design, the construct's secondary structure, physicochemical features, and allergenicity were predicted by SOPMA, Protparam, and AllergenFP, respectively. Then, the 3D structure was built via ITASSER. ProSa-web and PROCHECK were used for structure validation. The 3D model was docked by Cluspro, and molecular dynamics was carried out by GROMACS. Results: the results showed that the average RMSF value for the PSMA receptor was 0.478 Å, and for the designed toxin was 0.292 Å. The low range of changes in-dicates the stability of the complex during the simulation. Conclusion: The present results indicate that the designed immunotoxin is structurally stable, non-allergenic, and capable of binding PSMA, suggesting it as a potential candidate for further experimental evaluation.
Introduction :
Prostate Cancer (PCa) is the second most prevalent malignant tumor in men and the fifth leading cause of death worldwide 1. Although surgery or radiation therapy can successfully treat primary PCa, 18% of PCa patients in advanced stages die every year due to a lack of curative treatments 2,3. Therefore, the development of new molecular-targeted therapy methods seems as necessary as possible 4,5. A number of tumor cell components, such as glycoproteins or cell surface proteins that function as enzymes or receptors, are therapeutic targets for PCa treatment. The Prostate-Specific Membrane Antigen (PSMA) is commonly utilized as a therapeutic target for the generation of immunotoxin against PCa due to its constitutive or induced internalization property 6-8.
The PSMA as a type II membrane-glycoprotein with a molecular weight of 100 kDa is recognized as glutamate carboxypeptidase II or folate hydrolase, expressed in the prostate epithelial cells membrane 6. The PSMA structure is comprised of 750 residues, which can be separated into a small intracellular domain with 19 aa, a transmembrane domain with 21 aa, and a large extracellular domain with 707 aa 9, all of which is a compact 3D structure are expressed as a homodimer 7. In prostate cells, PSMA is involved in the uptake of cellular folate and glutamate that are primarily involved in DNA synthesis and repair, amino acid and polyamine generation, and the PI3K-Akt pathway 10.
It has recently attracted much attention in PCa therapeutic procedures due to its: (a) predominant expression in the prostate, (b) extensive expression at all stages of cancer like in androgen-insensitive or metastatic stage, (c) association with enzymatic or signaling activity, (d) presentation at the cell surface without releasing into the circulation and its capability to internalize after binding to antibody 2,9. Many monoclonal antibodies are specific for PSMA; the humanized monoclonal IgG1 antibody J591 is one of them. J591 is a 151 kDa antibody with high affinity and specific targeting in vivo. J591 binds to an external domain of PSMA, which can lead internalization of PSMA in prostate cancer cells 11-13.
Single chain variable-fragment (scFv) is one of the most popular antibody formats for tumor targeting 14. ScFv consists of the variable regions of the heavy chain (VH) and light chain (VL) 15 that are assembled by a peptide linker 16,17. The scFv fragments confer many advantages; the small size improves their penetration to tumor tissues and the capability of direct binding to toxins, which also intensifies the molecule stability of it in comparison with chemically coupled antibodies 2,18,19. Immunotoxins are proteins that consist of a toxin fused to an antibody or small molecules. The mode of action of immunotoxins is to kill designated cancer cells through the effector function of toxins. During the past several decades, the function of a wide range of immunotoxins on cancer cells has been studied, and the role of immunotoxins in the therapy of several kinds of cancers has been demonstrated 18,20-22.
Pseudomonas-Exotoxin (PE) A can be used as a single-chain toxin. It comprises 613 amino acids three structural domains that accelerate its function as a protein synthesis inhibitor in eukaryotic cells. The toxicity of PE without domain la to mice is almost 100-fold lower than intact PE, a feature that makes it a useful molecule for immunotoxins construction 23. PE40 is a truncated form of PE A that lacks domain 1a, which acts as a binding domain for cell surface receptors of most mammalian cells 23. PE40 does not show any toxicity as long as it remains in the extracellular space. Once an scFv is bound to direct it against a cell surface antigen and enable it to internalize, PE40 gets the immunotoxin potential 24.
Recombinant Immunotoxins (RITs) are a type of drug molecule that integrates antibody and protein toxin elements to significantly boost the ability of antibodies to kill cancer cells. At this time, a limited number of immunotoxin molecules have been sanctioned by the FDA for the clinical management of tumors. One such example is denileukin diftitox (Ontak; approved in 1999), which combines human interleukin-2 with truncated diphtheria toxin 25. Meng et al created a novel fusion combining an anti-Prostate-Specific Membrane Antigen (PSMA) antibody (J591) and diphtheria toxin (Fdt). The antitumor effects of immunocasp-3 were investigated in nude mice xenograft models featuring PSMA-overexpressing LNCaP cells 26.
Masilamani et al designed a PSMA-targeted, de-immunized PE24 variant. Preclinical studies indicated markedly better tumor uptake and survival benefits in prostate cancer models, highlighting endosomal escape strategies as a potential strategy for future immunotoxin therapies 27.
Nowadays, recombinant protein technology can produce proteins with desirable features, which makes a significant advancement 28-34. Furthermore, the bioinformatics approach accelerates analyzing biological data and designing novel biomolecules, which changed biology and medicine research 35-41. Here, various in silico methods are employed to design and evaluate a chimeric protein consisting of anti-PSMA mAbs (J591) and PE A fragments linked with J591 scFv.
Materials and Methods :
Construct design
An immunotoxin originating from an anti-PSMA scFv (J591) and a truncated form of Pseudomonas exotoxin A (PE40, aa 252-613, Uniprot ID: P11439) was designed to be incorporated as an efficient immunotoxin against PSMA for therapy purpose in prostate cancer. The binding part of the construct was constituted by fusing the antibody fragments (VL-VH of J591) using a flexible connector (GSTSGGGSGGGSGGG GSS). Furthermore, the PE40 and J591 antibody-derived scFv were correlated by the GSTSGGGSGGG SGGGGSS sequence as an accurate linker (Figure 1). Thus, the immunotoxin is composed of an anti-PSMA scFv as binding-domain and a truncated form of Pseudomonas aeruginosa-Exotoxin A (PE40) as toxin-domain.
Secondary structure features
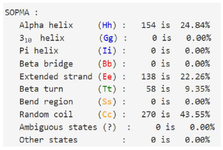
The secondary structure of the J591 antibody-derived scFvPE40 fusion form was submitted in the SOPMA server 42. This server predicted the secondary structural properties and 4 conformational states (Helix, Sheet, Turn, and Coil) of the final structure.
Physicochemical properties
ProtParam as an online accessible web server was utilized for evaluation of several physicochemical parameters of the designed immunotoxin sequence (including theoretical isoelectric point (pI), Molecular Weight (MW), aliphatic index, instability index, grand average of hydropathicity (GRAVY) and half-life) 43.
Allergenicity
The potential allergenicity of designed immunotoxin (whole fusion protein) was examined by two bioinformatics online servers known as AllergenFP v.1.0 and AllerTOP v.2.0. AllergenFP v.1.0 is according to a new alignment-free descriptor-based fingerprint procedure 44. The advantage for the AllerTOP v.2.0 server is using Auto Cross-Covariance (ACC) mode and numerous allergens and non-allergens from varied species have also been used to train it 45.
mRNA stability
RNAfold web server, based on energy minimization, was used to consider the mRNA secondary structure of designed immunotoxin. The encoding gene was optimized for expression in bacterial host.
3D structure estimation, refinement, and validation
AIterative Threading ASSEmbly Refinement server (I-TASSER) was utilized 46, as a freely available online web server, widely used for the prediction of the three-dimensional structures of the designed immunotoxin. This server predicts the structure and function of the protein by a hierarchical approach. At first, according to the high C-score, the best 3D structure is selected. Then, GalaxyRefine server 47 was used to approve the preferred structure and for further improvement. Two freely available web tools were employed for analyzing and validation of the selected 3D-models, in two steps before and after improvement. ProSA-web 48 verified errors in the 3D-structures via computing a total quality score (Z-score). If the estimated Z-score of the preferred 3D-structure is away of the characteristic range for native proteins, likely indicates structural errors. PROCHECK server 49 was employed for validation of 3D models through the Ramachandran plot. Finally, the residues were categorized into three regions, i.e., favored, allowed, and disallowed regions.
Molecular docking of PSMA receptor with designed immunotoxin
To evaluate the interaction of the designed immunotoxin as a ligand with PSMA as a receptor, molecular docking analysis using Cluspro web server was done 50 and the default parameters of the server were exerted.
Simulation of molecular dynamics
To evaluate the stability of the strongest interaction of designed immunotoxin-PSMA complex, MD simulation was performed using the GROMACS 2018 package 51 and OPLS-AA force field 52. OPLS/AA force fields have been widely used for the simulation of protein and protein modification of organic molecules with many different functional groups. This system allows faster calculations to evaluate the stability of protein-protein interactions. OPLS/AA force field was used for studying the stability and their changes through interaction immunotoxin with PSMA. The complex soaked in a solvent box with a minimum distance of 1.0 nm between the solutes’ surface and the box. The box was filled with TIP3P explicating water solvent 53. Instead of solvent molecules, Na+ and Cl- ions in the simulation box were used to neutralize the charge of the whole system. For the MD simulation process, the energy minimization consisted of two parts: first, the system using NVT (constant Number of particles-Volume-Temperature) was equilibrated at 300 K for 100 ps and second, the device was equilibrated using a Parrinello-Rahman barostat to get fixt temperature and pressure at 300 K and 1.0 bar. Van-der-Waals (VDW) interactions were computed with a 1 nm cutoff. The Linear constraint (LINCS) algorithm was utilized to restrict the length of covalent bonds. Following necessary equilibrations, 100 ns production runs were carried out for designed toxin-PSMA complex. The stability of the designed immunotoxin-PSMA complex was measured by evaluating the Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), and Radius of Gyration (RG) parameters.
Results :
Secondary structure features
The SOPMA server was employed to assess the secondary structure of the designed immunotoxin (Figure 2). The percent of predicted conformational states include 24.84% alpha-helical regions, 22.26% extended-strands, 9.35% beta-turns, and 43.55% random-coil.
Physicochemical properties and allergenicity of the designed immunotoxin
ProtParam server determined the physicochemical trait parameters of the construct (Table 1). This analysis showed the designed immunotoxin is an acidic, negatively charged, stable protein. The both AllerTOP v.2.0, and AllergenFP v.1.0 servers were utilized to predict allergenicity (Table 1). These servers predicted that scFv+PE40 construct could be a non-allergenic protein and this represents that the immunotoxin does not cause inflammatory responses and allergic reaction in human body.
mRNA constancy
RNAfold online software was utilized to evaluate the free energy minimization and be assured of the probable stability of the chimeric mRNA. -916.68 kcal/mol was recorded for the free energy of the thermodynamic ensemble, as the secondary structure of the mRNA construct is illustrated in figure 3.
3D model construction, clarification, and validation
I-TASSER server built five three-dimensional structures for the chimeric protein sequence, among them the finest structure with the highest C-score (-1.02) was selected (Figure 4A). The 3D structure of the selected model was refined using the GalaxyRefine server, and information on the structural changes from the initial structure is provided in terms of RMSD, MolProbity, Ramachandran plot, GDT-HA, Poor rotamers, and clash score (Figure 4C). The constructed model that had the most favored region in the Ramachandran plot was selected (Figure 4B). Model 3 was considered as the best-refined model.
Two servers consisting ProSA-web and PROCHECK were applied to determine the quality level and possible errors in the preferred model of the prior step. The validation of the refined structure by the PROCHECK verified that 86.9% of residues are in the recommended region, 10.0% in the permitted region while only 2.2% in the disallowed region. Whereas in the primary structure, Ramachandran plot analysis represented that 73.5, 20.7, and 2.2% of residues were included in the favored, permitted, and disallowed regions, respectively. The Ramachandran plots obtained for primary and refined structures are given in figure 5.
To further determine the quality of the model, the ProSA-web was applied and based on the results of ProSA-web, the Z-score of the original model was distinguished as −8.94. To further improving this model; after all refinements, the refined model reached Z-score of −9.09 (Figure 6).
Molecular docking of PSMA receptor with designed immunotoxin
3D models for interaction between the PSMA receptor and designed immunotoxin ligand were created using the Cluspro web server. In results, the balanced coefficients were used. The well-organized-predicted model for ligand and receptor docking with the lowest energy score is shown in figure 7.
Simulation of molecular dynamics
The interactions of the designed toxin-PSMA complex were selected for MD simulation for 100 ns. The stability of the docked complex was analyzed using the RMSD, RG, RMSF, and SASA values. The average RMSD values of the designed toxin-PSMA complex were 0.45 nm. As shown in figure 8A the complex reached stability after about 40 ns. Moreover, the Rg parameter was evaluated for determining the degree of compression and the size of protein molecules. The mean Rg values of the designed toxin-PSMA complex were calculated to be approximately 3.66 nm. The changes in Rg values of the complex during the simulation were between 3.5 nm and 3.7 nm, so it can be concluded that the compactness of the complex is stable after about 40 ns the simulation (Figure 8B). RMSF values are a measure of atomic fluctuations along the MD simulation. Analysis of the results showed that the average RMSF value for the PSMA receptor (Figure 9A) was 0.478 Å, and for the designed toxin (Figure 9B) was 0.292 Å. The low range of changes indicates the stability of the complex during the simulation.
Discussion :
Immunotoxin molecules have much potential in cancer treatment, and several have already been approved for clinical trials 54. PE (Pseudomonas exotoxin A) is a highly toxic poison that acts as the killing moiety in a number of immunotoxins 55. Compared to other toxins, PE is a promising toxin for developing RITs according to its great potency, expression, and purification profits, simple cloning feature, and low non-specific toxicity 56,57. PE40 is a truncated form of PEA; it misses the cell-binding domain but retains domains II and III, which are comprised of membrane translocation and inhibition of protein synthesis in eukaryotic cells 58,59.
In the current study, a chimeric protein was designed and tested using various in-silico tools, including the exotoxin from Pseudomonas A fragment associated with anti-PSMA J591 scFv. To begin designing an immunotoxin against prostate cancer, protein sequences of J591 scFv and Pseudomonas exotoxin PE40 were obtained. PE40 is an efficient toxin that is studied in various immunotoxins. Furthermore, J951 scFv is a specific PSMA targeting molecule. PE40-J951 fusion could be an efficient immunotoxin for prostate cancer. Then, the construct's components were assembled using a flexible linker (GSTSGGGSGGGSGGGGSS). The SOPMA server was used to consider the secondary structure of the designed immunotoxin.
The ProtParam online web server investigated the immunotoxin's physicochemical properties (MW, aliphatic index, instability index, etc.). The theoretical pI highlighted the essential characteristics of the protein, offering advantages for methodologies such as ion-exchange chromatography and isoelectric focusing. The protein displayed a borderline instability index of 38.08. It is vital to stress that an instability index below 40 is typically regarded as stable. Moreover, the GRAVY score for the construct was calculated to be -0.379, indicating hydrophilicity and illuminating the protein's solubility; the negative score points to advantageous interactions with water molecules. In addition, the notable aliphatic index of 71.97 suggests the possible thermodynamic stability of the modified protein 60.
The analysis revealed that the designed immunotoxin is a stable, negatively charged protein. Allergen and AllerTOP servers were used to assess the potential allergenicity of the designed immunotoxin. According to the results obtained from these servers, the designed structure can be considered a non-allergenic protein that does not stimulate inflammatory responses in the human body. A prominent limitation of immunotoxins lies in their significant immunogenicity, primarily because their cytotoxic components are often sourced from bacterial proteins such as Pseudomonas exotoxin A (PE). In patients with intact immune systems—particularly those with solid tumors—this foreign aspect typically incites vigorous humoral responses, culminating in the rapid development of anti-drug neutralizing antibodies that drastically reduce therapeutic exposure and effectiveness over successive treatment cycles 61.
RNAfold online software was used to assess the free energy minimization for predicting the probable stability of the chimeric mRNA. The negative free energy values indicated the mRNA construct's secondary structure stability. The I-TASSER server was employed to anticipate the 3-dimensional constructions of the designed immunotoxin. The best 3D structure is chosen based on the high C-score. With further development, the 3D structure of the best model was refined by using the GalaxyRefine server. The selected 3D models were analyzed and validated using ProSA-web and the PROCHECK server. Model 3 was thought to be the most refined model, with the best Ramachandran plot region.
Molecular docking and molecular dynamics simulation analyses yield important insights regarding the binding stability and structural dynamics of the developed immunotoxin–PSMA complex. Similar investigations aimed at PSMA have applied docking followed by molecular dynamics simulation to assess ligand–protein interactions. As an example, Nikfarjam et al executed molecular docking and molecular dynamics simulations on PSMA with small-molecule compounds, illustrating how these combined computational techniques can identify strong inhibitors and analyze stability in relation to docking poses and dynamic behavior 62.
The molecular dynamics simulation performed over a duration of 100 ns validated the stability of the docked complex. The analysis of RMSD indicated that the immunotoxin–PSMA complex reached equilibrium after around 40 ns and maintained stability with an average RMSD of 0.45 nm. This finding aligns with standard methodologies in molecular dynamics studies, where RMSD plots are utilized to illustrate the stability of protein–ligand complexes over time 63. Although not explicitly focused on PSMA, other molecular dynamics studies have documented RMSD convergence and stability values that are within similar ranges, thus bolstering the trustworthiness of this stability assessment.
The values of the Rg, averaging around 3.66 nm and ranging from 3.5 to 3.7 nm, reflect a stable compactness and structural integrity of the complex, which stabilizes after approximately 40 ns. This finding corresponds with conventional interpretations in molecular dynamics simulations, where Rg is utilized to measure conformational compactness and its temporal stability 64.
The RMSF analysis revealed low fluctuations for both the receptor (0.478 Å) and the immunotoxin (0.292 Å), which suggests that there are minimal conformational changes and a stable binding interface. In molecular dynamics studies of other systems, low RMSF values have similarly been observed as indicators of strong protein–ligand stability; for example, residue fluctuations under ~0.3 nm (i.e., 3 Å) indicate high backbone stability 65.
Overall, the consistent patterns observed in structural parameters—RMSD equilibrium, stable Rg, and minimal fluctuations in RMSF—underscore the robust and stable nature of the designed immunotoxin–PSMA interaction. These computational results are consistent with the broader literature that illustrates how low dynamic variability and compact conformations are indicative of effective ligand binding and structural compatibility 66.
Conclusion :
The current study attempted to design an effective immunotoxin for the treatment of prostate cancer using an in silico strategy. We designed, evaluated, and selected the immunotoxin structure using various bioinformatics tools and servers. Besides that, docking and molecular dynamics studies were used to determine the immunotoxin's affinity for PSMA. The designed immunotoxin had a stable structure based on the results. Moreover, the ligand-receptor binding study indicates that the immunotoxin can bind to the PSMA successfully.
Ethical approval
All procedures conducted in this research were approved by ethical committees of Behbahan Faculty of Medical Sciences (IR.BHN.REC.1401.007).
Conflict of Interest :
The authors declare that there is no conflict of interests.
Funding: This work is financially supported by Behbahan Faculty of Medical Sciences, Behbahan, Iran (Grant number: 400060).
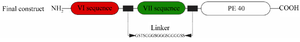
Figure 1. Schematic presentation of the final designed immunotoxin.
|
|

Figure 3. The free energy of the thermodynamic ensemble is -916.68 kcal/mol.
|
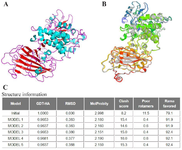
Figure 4. (A) Predicted 3D structure for the fusion protein by I-TASSER server. The 5 best models (B) and structure information (C) as outputs for Galaxy Refine server, are shown.
|
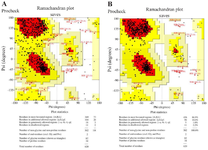
Figure 5. PROCHECK analysis for the primary and refined structures. A) In the primary structure 73.5, 20.7, and 2.2% of residues were in favored, allowed, and disallowed regions, respectively. B) In the refined structure 86.9, 10.0, and 2.2% of residues were in favored, allowed, and disallowed regions, respectively.
|
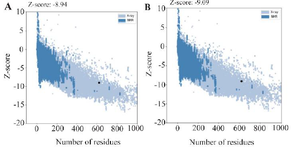
Figure 6. The Z-scores predicted by the PROSA-web server for initial and refinement structure are given in panel (A) and (B), respectively.
|
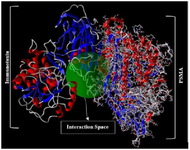
Figure 7. Molecular docking of anti-PSMA scFv as a ligand with PSMA receptor. The best results of docking interactions between ligand and receptor with the lowest energy score (-1135.7 kcal/mol) presented by this model.
|
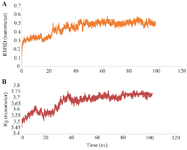
Figure. 8 A) The RMSD values designed toxin-PSMA complex. B) The Rg values of the designed toxin-PSMA complex.
|
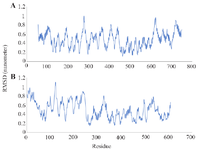
Figure 9. The RMSF values designed toxin-PSMA complex. A) The RMSF values of PSMA in complex form. B) The RMSF values of designed toxin in complex form.
|
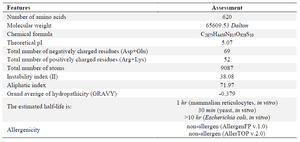
Table 1. Summarize of the physicochemical parameters, allergenicity of the designed immunotoxin
|
|