Expression, Purification, and Application of SARS-CoV-2 Nucleocapsid Protein for Serological Detection of IgG and IgM Antibodies
-
Moosavi, Farshid
-
Monoclonal Antibody Research Center, Avicenna Research Institute (ACECR), Tehran, Iran
-
Zamani, Bahareh
-
Monoclonal Antibody Research Center, Avicenna Research Institute (ACECR), Tehran, Iran
-
Agharezaee, Niloofar
-
Monoclonal Antibody Research Center, Avicenna Research Institute (ACECR), Tehran, Iran
-
Yousefi, Parisa
-
Monoclonal Antibody Research Center, Avicenna Research Institute (ACECR), Tehran, Iran
-
 Nazari, Mahboobeh
Monoclonal Antibody Research Center, Avicenna Research Institute (ACECR), Tehran, Iran, Tel: +98 21 22432020; Fax: +98 2122432021; E-mail: nazari1980azar@yahoo.com
Nazari, Mahboobeh
Monoclonal Antibody Research Center, Avicenna Research Institute (ACECR), Tehran, Iran, Tel: +98 21 22432020; Fax: +98 2122432021; E-mail: nazari1980azar@yahoo.com
Abstract: Background: SARS-CoV-2 is a novel coronavirus that has caused dramatic loss of life and poses an unprecedented public health challenge worldwide. The nucleocapsid (N) protein of SARS-CoV-2 is the most abundant viral protein and a potent immunogen. Methods: In the current study, the N gene of SARS-CoV-2 was amplified from RNA extracted from a COVID-19 positive patient and then cloned into the pCold-I expression vector. The full-length His-tagged N protein was expressed in Escherichia coli (E. coli) using 0.5 mM IPTG and subsequently purified by nickel affinity chromatography. The purified N protein was characterized using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting. Furthermore, the purified N protein was applied in SARS-CoV-2 IgG and IgM ELISA immunoassays. Results: The results showed that purification of the N protein in the presence of urea yielded a protein band of approximately 48 kDa on SDS-PAGE, corresponding to the full-length N protein. Additionally, Western blot analysis of the purified recombinant N protein showed a band of the same molecular weight. In the SARS-CoV-2 IgG ELISA assay, anti-N protein antibodies from a COVID-19 positive patient’s serum successfully recognized the coated N protein. In the IgM ELISA test, an N-HRP conjugate was used in ELISA wells to reveal the interaction of HRP-conjugated N protein with pre-coated anti-N protein antibodies (IgM isotype). Conclusion: These results indicate that the expressed N protein of SARS-CoV-2 could serve as a valuable reagent for the development of antibody-based immunoassays to detect SARS-CoV-2 IgG and IgM antibodies.
Introduction :
In December 2019, Coronavirus disease 2019 (COVID-19) was reported from China. The coronavirus disease was classified on March 12, 2020, as a global pandemic 1. Later, the International Committee on Taxonomy of Viruses (ICTV) designated this disease as SARS-CoV-2. Similar to SARS coronaviruses, SARS-CoV-2 can cause severe respiratory disease, and people over 60 years old suffer significant mortality 2.
Structural SARS-CoV-2 proteins include Spike protein (S), Envelope protein (E), Membrane protein (M), and Nucleocapsid protein (N). The N protein, as the nuclear localization signal, has 419 amino acids which plays a significant role in the virus particle assembling 3. The SARS-CoV-2 N protein has five regions including a predicted N-terminal section (1–40 aa), N-terminal domain (NTD 41–186 aa), predicted central linker (187–257 aa), C-terminal domain (CTD, 258–361 aa), and a predicted C-terminal domain (362–419 aa) 4. It has been revealed that the N protein accumulates intra-cellularly and also it is one of the virus antigens highly produced throughout the infection process. The N Protein is a relatively conserved protein in coronaviruses, and hence it could be as a strong immunogen in several coronaviruses; as a result, it has been frequently used as a protein candidate in the development of serological assays 5. Additionally, in different RNA viruses such as mumps, rabies, vesicular stomatitis, measles, Newcastle disease, and IBV viruses, the N proteins have been applied as suitable antigens in diagnostic immunoassay tests.
The production of accurate diagnostic tests is a critical tool for controlling outbreaks. Hence, the production of SARS-CoV-2 antigens is an important occasion to develop reliable serological assays. For a sensitive and reliable serological assay against SARS-CoV-2, it is necessary to produce SARS-CoV-2 N protein. Protein expression in prokaryotic systems, such as Escherichia coli (E. coli), is a cost-effective way to provide high quantities of recombinant proteins 6.
Upon coronavirus infection (3−6 days), the IgM antibodies are produced by short-lived plasma cells during the early phase of the B-cell response, providing the first line of adaptive defense against viral infections, whereas the long-term humoral response is based on high-affinity IgG, which could be detected after 8 days and provides information on the time-course of virus infection. Therefore, the detection of both IgM and IgG antibodies could provide information for confirming SARS-CoV-2 infection in the suspected patients 7. In this study, in order to explore the accurate and reliable detection for COVID-19 diagnosis, the SARS-CoV-2 N protein gene was cloned and expressed in an E. coli strain and then purified. Subsequently two ELISA assays using recombinant N Protein were developed as the diagnostic target and assessed its performance for the clinical diagnosis of SARSCoV-2 infections by detecting N protein-specific IgM and IgG antibodies in patients 8.
Materials and Methods :
The cloning of SARS-CoV-2 N gene
The RNA of the SARS-CoV-2 virus was extracted from the throat sample of a positive COVID-19 patient using Ambion PureLink RNA Mini Kit (Thermo Scientific, Waltham, Massachusetts, USA) according to the manufacturer’s recommendations. RNA integrity was confirmed by agarose gel electrophoresis and the concentration was determined by measuring the Optical Density (OD) at 260 nm using a NanoDrop spectrophotometer (Thermo Scientific, Waltham, Massachusetts, USA). DNA contamination was removed using a commercial kit (Sigma, Missouri, USA, Product Number: AMPD1) according to the manufacturer’s recommendation. First-strand cDNA was synthesized according to the manufacturer’s protocol (Thermo Scientific-K1621) as follows: five μg (10 μl) of the DNA-decontaminated RNA (Final RNA concentration = 0.25 µg/µl), 4 μl 5× reaction buffer, 2 μl dNTPs, 1 μl N6 random hexamers, 1 μl reverse transcriptase, and 2 μl water in a total volume of 20 μl. The reaction was performed as follows: 5 min at 25°C, 60 min at 42°C, and 5 min at 70°C. The synthesized cDNA was stored at -20°C until further use.
A 1276 bp PCR product was amplified using sense 5'- TGGATCCTGTGTTAATCTTACAACCAGAACT C-3' and antisense 5'- TTTAAGCTTAAGAATTAGTCTGAGTCTGATAACTAGC -3' primers. A PCR program was used for DNA amplification as follows: 94°C for 5 min, a 30-cycle amplification (98°C for 10 s, 55°C for 30 s, and 72°C for 30 s), and a final extension at 72°C for 5 min, using a Mastercycler Gradient Eppendorf Thermocycler. PCR reactions were performed in a 20 μl volume containing 2 µl cDNA, 0.25 µl (10 pmoles/µl) of each primer (Final concentration = 0.125 µM), 7.5 µl water, and 10 µl Taq DNA Polymerase Master Mix RED (Ampliqon, Danmark). The expected PCR product size, 1276 bp, for the SARS-CoV-2 N gene was evaluated by 1% agarose gel electrophoresis. Amplicons were double-digested by NdeI/ BamH1 and inserted into the digested/dephosphorylated pCold-I (Takara Bio, Mountain View, CA, USA) expression vector. The ligated mixtures were chemically transformed into E. coli DH5α. Positive colonies were screened using a colony PCR experiment. Finally, plasmids were extracted and further confirmed through double digestion and sequencing experiments.
Expression and purification of the recombinant N protein
The constructed plasmid was chemically transformed into E. coli strain Gold BL21 (DE3) (Novagen, Wisconsin, USA) and subsequently cultivated in 3 ml of Luria-Bertani (LB) broth containing ampicillin (100 µg/ml) with shaking (220 rpm) overnight at 37°C. The overnight culture was then transferred into 300 ml of fresh LB broth containing ampicillin. When the OD at 600 nm (OD₆₀₀) reached 0.9, protein expression was induced by adding 0.5 mM IPTG and incubating overnight at 15°C. Cells were harvested after 24 hr by centrifugation at 4500 rpm for 20 min and lysed in lysis buffer containing 50 mM Tris, 500 mM NaCl, 10 mM imidazole, and 8 M urea, pH=8, using sonication. The soluble fraction was separated by centrifugation (30 min at 10,000 rpm) and applied to a nickel ion affinity column (GE Healthcare). The column was washed with five volumes of wash buffer (50 mM Tris, 500 mM NaCl, 50 mM imidazole, and 8 M urea, pH=8). The protein was eluted with elution buffer containing 50 mM Tris, 500 mM NaCl, 500 mM imidazole, and 8 M urea, pH=8.
Characterization of the purified N protein using SDS-PAGE and western blot
The purified proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) on a 10% gel. Specifically, the purified proteins were solubilized in sample buffer containing 62.5 mM Tris–HCl (pH=6.8), 1% SDS, 10% glycerol, 0.001% bromophenol blue, and boiled for 5 min. After electrophoresis, protein bands were visualized by Coomassie Brilliant Blue R-250 staining. For western blotting, the separated proteins were transferred onto a PVDF mem-brane at 200 mA for 2 hr. The membrane was blocked with 5% skim milk overnight at 4°C, then incubated with mouse anti-His-tag antibody as the primary antibody (Padzaco, Tehran, Iran), followed by incubation with HRP-labeled sheep anti-mouse antibody (Avicenna Research Institute, Tehran, Iran). After antibody incubation, the membrane was washed twice with TBS supplemented with Tween 20. The protein bands were finally visualized using enhanced chemiluminescence (ECL) (Millipore Corporation, Billerica, MA 01821).
SARS-CoV-2 IgG ELISA assay
Strips were coated with recombinant N protein at a concentration of 1 μg/ml in Phosphate-Buffered Saline (PBS) (pH=7) at 4°C overnight. The strips were then washed three times with PBS containing 0.05% Tween 20 and incubated with 5% Bovine Serum Albumin (BSA) for 2 hr at 37°C. After washing, human sera were diluted 1:100 and incubated for 30 min at 37°C. Following additional washes, HRP-conjugated goat anti-human IgG (Sigma-Aldrich, St. Louis, MO, USA), diluted 1:30,000, was added and incubated for 30 min at 37°C. Finally, 3,3′,5,5′-tetramethylbenzidine (TMB) was added to develop a blue color, and the reaction was stopped with 20% H₂SO₄. The OD was measured at 450 nm using an ELISA reader (Convergys EL-Reader96X).
N protein–HRP conjugation
The purified N protein was conjugated to HRP using the periodate method. Specifically, the N protein (4 mg/ml) was dialyzed against 1 M NaCl at 4°C overnight. HRP (Biobasic, Toronto, Canada) was dissolved in water (5 mg/ml) and oxidized using 10 mM sodium periodate for 20 min at Room Temperature (RT) in the dark with gentle shaking. The oxidized HRP was immediately dialyzed against 1 mM sodium acetate buffer (pH=4.4) at 4°C overnight. Then, 5 mg of oxidized HRP was mixed with 1 mg of N protein, and the pH was adjusted to 9.2 using 100 mM bicarbonate buffer. The mixture was incubated for 2 hr at RT. Next, 50 mM sodium cyanoborohydride was added and incubated for an additional 2 hr at 4°C. The N protein–HRP conjugate was then dialyzed against PBS (pH=7.2) at 4°C overnight.
SARS-CoV-2 IgM ELISA assay
In this ELISA, strips were coated with anti-human IgM polyclonal antibody (Sina Biotech, Tehran, Iran) at 5 μg/ml in PBS (pH=7) at 4°C overnight. The strips were washed with PBS containing 0.05% Tween 20 and then blocked with 5% BSA for 2 hr at 37 °C. After washing, human sera were diluted 1:3 and incubated for 30 min at 37°C. Following additional washes, N protein–HRP conjugate (diluted 1:500) was added and incubated for 30 min at 37°C. Finally, TMB substrate was added to develop a blue color, and the reaction was stopped using 20% H₂SO₄. The OD was measured at 450 nm using an ELISA reader (Convergys EL-Reader96X).
Results :
Cloning of the SARS-CoV-2 N gene into the pCold-I vector
Viral RNA was extracted from the throat swab of a COVID-19-positive patient and examined using a spectrophotometer at wavelengths ranging from 220 to 330 nm (Figure 1A). The quality of the extracted RNA was assessed by electrophoresis on a 1.2% agarose gel, where RNA bands (28S, 18S, and 5S) were visualized (Figure 1B). After synthesizing cDNA from the extracted RNA, a housekeeping gene (human GAPDH) was used to evaluate the quality of the synthesized cDNA. Using specific primers targeting the SARS-CoV-2 N gene, the desired amplicon was successfully amplified (Figure 1C). The PCR product was then cloned into the pCold-I plasmid. Restriction enzyme digestion of the recombinant plasmid confirmed the presence of the insert by releasing two DNA fragments corresponding to the insert and the vector, 1276 bp and 3000 bp, respectively (Figure 1D). Finally, the SARS-CoV-2 N gene was confirmed by sequencing.
Expression and purification of the recombinant N protein
The SARS-CoV-2 N protein, containing a C-terminal His-tag, was expressed in E. coli strain Gold BL21 (DE3) and purified using nickel ion affinity chromatography under both denaturing and non-d enaturing conditions. Protein expression was induced with 0.5 mM IPTG. After 24 hr, the concentration of N protein in cell lysates was assessed under denaturing conditions (8 M urea) and non-denaturing conditions. SDS-PAGE analysis (Figure 2A) showed a higher yield of N protein under denaturing conditions (lane 1) compared to non-denaturing conditions (lane 2). No N protein band was detected in the uninduced condition (lane 3). During purification, N protein was eluted in the presence of 8 M urea and subsequently dialyzed against PBS containing 10% glycerol to promote protein refolding. After refolding, a protein band of approximately 48 kDa was observed by SDS-PAGE (Figure 2B). Western blot analysis revealed that this band was specifically recognized by an anti-His tag antibody, confirming the expression of the recombinant N protein (Figure 2C).
N protein–HRP conjugation
The N protein was conjugated to HRP for use in the SARS-CoV-2 IgM ELISA assay. As shown in figure 3, SDS-PAGE analysis revealed the appearance of new protein bands with higher molecular weights after conjugation. The molecular weights of the native N protein and HRP are approximately 48 kDa and 44 kDa, respectively. Following conjugation, the emergence of smeared high molecular weight bands, relative to intact N and HRP, indicated successful conjugation of the N protein to HRP molecules.
Reactivity of the N protein for detection of SARS-CoV-2 antibodies by ELISA
The reactivity of the purified N protein was evaluated using sera from COVID-19 patients in two ELISA platforms: SARS-CoV-2 N protein IgG and IgM ELISA tests (Figures 4A and B). In the IgG ELISA, the N protein was coated on ELISA wells, followed by incubation with patient serum containing IgG antibodies. As shown in figure 4A, the anti-N protein IgG antibodies in patient serum were able to recognize the coated N protein at various dilutions. Serum from a healthy individual served as a negative control. In the IgM ELISA, anti-human IgM antibodies were first coated onto ELISA wells to capture serum IgM. Subsequently, the N protein–HRP conjugate was added in different dilutions to detect binding to human IgM anti-N protein antibodies (Figure 4B). The results showed that the HRP-labeled N protein effectively bound to the IgM antibodies. A healthy serum was again used as a negative control.
Discussion :
Since the emergence of the SARS-CoV-2 virus in 2019, researchers have made significant efforts to develop rapid and accurate diagnostic methods for its early detection. Several published studies have shown that anti-N protein antibodies present in patient sera can be used to confirm SARS-CoV-2 infection using ELISA platforms 9. In this context, the current study aimed to clone, express, and characterize the full-length recombinant SARS-CoV-2 N protein in E. coli to evaluate its potential for use in ELISA-based detection systems 10.
In this study, the full-length N protein was successfully engineered, expressed in E. coli, and purified from the cell lysate supernatant in the form of inclusion bodies. Consistent with previous findings, Liang et al reported that their full-length recombinant N protein expressed in E. coli also formed insoluble precipitates 11. Here, in this study it was demonstrated that the urea-denatured full-length N protein could be successfully refolded into its functional state by buffer exchange from urea-containing buffer to PBS.
Importantly, the N protein was purified using a Ni-NTA affinity column facilitated by a C-terminal His-tag. Since anti-His-tag antibodies are not present in human sera, the presence of the His-tag does not interfere with immunoassays 12. Some commercial ELISA kits use viral protein mixtures obtained through the cultivation of SARS-CoV-2 13. However, preparing such mixtures is a laborious and complex process. As a simpler and more practical alternative, several studies have proposed the use of purified N protein as a specific, sensitive, and easily producible antigen for ELISA-based detection of SARS-CoV-2 antibodies 14,15. More-over, the N protein has been reported to be more immunogenic than other SARS-CoV-2 proteins 16.
Conclusion :
Both IgM and IgG antibodies play crucial roles in antiviral immune responses. IgM antibodies typically appear in the early stages of infection, while IgG antibodies emerge later and can provide long-term immunity 17,18. Therefore, in this study, both IgG and IgM ELISA assays were designed and developed to detect anti-N protein antibodies in patient sera, enabling comprehensive serological detection across different stages of infection.
Ethical approval
All procedures conducted in this research were approved by ethical committees of Avicenna Research Institute (IR.ACECR.AVICENNA.REC.1399.024).
Acknowledgement :
The authors are thankful to the staff of Avicenna Research institute.
Conflict of Interest :
The authors declare no conflict of interest.
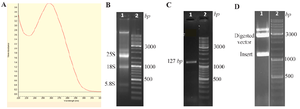
Figure 1. Cloning of the SARS-CoV-2 N gene.
A) Viral RNA extraction was confirmed using UV–visible spectrophotometry in the range of 220–330 nm.
B) The quality of the extracted RNA was verified by agarose gel electrophoresis; lane 1: RNA showing 28S, 18S, and 5.8S bands; lane 2: molecular weight ladder.
C) RT-PCR amplification of the N gene; lane 1: amplicon of the N gene (1276 bp); lane 2: ladder.
D) Double digestion of the recombinant plasmid; lane 1: released N gene insert (1276 bp) and digested vector backbone (3000 bp); lane 2: ladder.
|
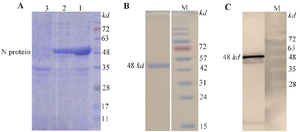
Figure 2. Expression and purification of the recombinant N protein.
- A) Expression of N protein under denaturing conditions (8 M urea, lane 1) and non-denaturing conditions (without urea, lane 2) assessed by SDS-PAGE; lane 3: uninduced sample showing no N protein expression.
- B) Purified N protein under denaturing conditions revealed a distinct band at ~48 kDa on SDS-PAGE.
- C) Western blot analysis detected a 48 kDa band using anti-His tag monoclonal antibody, confirming expression of the recombinant N protein.
|
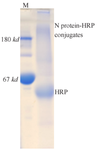
Figure 3. Conjugation of N protein to HRP. SDS-PAGE analysis showed the appearance of smeared bands with high molecular weights (>180 kDa), indicating successful conjugation of N protein (48 kDa) to HRP (44 kDa).
|
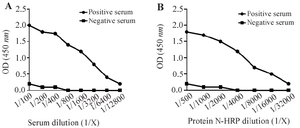
Figure 4. Reactivity of the N protein with SARS-CoV-2 antibodies in ELISA.
- A) In the IgG ELISA, anti-N protein antibodies in a COVID-19 patient’s serum recognized the coated N protein. A healthy serum was used as a negative control.
- B) In the IgM ELISA, HRP-conjugated N protein was applied to wells containing captured IgM antibodies from patient serum. The interaction confirmed specific binding. A healthy serum was used as a negative control.
|
|