Comparison of CRISPR Sequences in Archaea and Bacteria with Eukaryotic microRNAs
-
 Behbahani, Mandana
Faculty of Biotechnology, Isfahan University, Isfahan, Iran, Tel: +98 31 37934327, Fax: +98 31 37932456; E-mail: ma_behbahani@yahoo.com
Behbahani, Mandana
Faculty of Biotechnology, Isfahan University, Isfahan, Iran, Tel: +98 31 37934327, Fax: +98 31 37932456; E-mail: ma_behbahani@yahoo.com
-
Hejazi, Fatemeh
-
Faculty of Nanochemical Engineering, Shiraz University, Shiraz, Iran
Abstract: Background: This study explores repetitive Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) sequences from the archaea Acidianus sp. and Acidianus ambivalens (A. ambivalens), as well as from the bacterium Yersinia ruckeri (Y. ruckeri). These sequences are compared with human microRNA (miRNA) sequences to investigate potential genetic similarities and disease associations.
Methods: CRISPR sequences were retrieved from the CRISPR/Cas++ database, and human miRNA sequences were obtained from miRBase. Sequence alignments were performed using BLASTn with an E-value threshold of 1e-5 to identify significant similarities. Genes associated with matched human miRNAs were identified through the HGNC and GeneCards databases. Further analyses included comparison with disease-associated miRNAs reported in human and mouse datasets.
Results: In Y. ruckeri, alignments revealed similarities to miRNAs linked with genes such as FOXO1, PTEN, PAX7, and DOCK3, which are associated with lung cancer and muscular dystrophies. In A. ambivalens, aligned miRNAs corresponded to loci including CHM13 and GRCh38, potentially linked to periembolic adenocarcinoma and mild pre-eclampsia. For Acidianus sp., matches were observed with miRNAs associated with genes like Irak2, NOS2, STAT1, and Numb, which have been implicated in Psoriatic arthritis, Alzheimer’s disease, Hepatocellular carcinoma, and Coronary artery disease.
Conclusion: CRISPR sequences from these prokaryotes show notable similarities with human miRNAs, suggesting possible indirect links to genes involved in major diseases. These preliminary findings emphasize the need for further investigation into shared sequence motifs and their functional roles in host-pathogen interactions or evolutionary biology.
Introduction :
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and their associated Cas proteins constitute an adaptive immune system in bacteria and archaea, providing defense against mobile genetic elements such as phages and plasmids 1-4. Since their discovery in Escherichia coli (E. coli) by Ishino et al in 1987 5 (Figure 1), CRISPR/Cas systems have collected significant scientific interest due to their configurable functionality and broad applications in genome editing and molecular biology.
Despite extensive research into the classification, structure, and function of CRISPR arrays in prokary-otes, limited knowledge exists regarding the presence or potential analogs of these systems in eukaryotic microorganisms. The current body of literature has largely overlooked the possibility of CRISPR-like repetitive elements in unicellular eukaryotes, leaving a critical gap in our understanding of their evolutionary and functional relevance 6-9.
This study seeks to address this gap by systematically comparing CRISPR repeat sequences from bacterial and archaeal genomes with sequence elements identified in eukaryotic microorganisms. We hypothesize that specific repetitive motifs or structural analogs may be present in eukaryotic genomes, potentially shedding light on the evolutionary origins of CRISPR-like systems.
To explore this hypothesis, CRISPR sequences were obtained from the CRISPR/Cas++ database, while human miRNA sequences were retrieved from the miRBase repository. Sequence alignments were performed using BLASTn with an E-value threshold of 1e-5 to detect statistically significant similarities. These computational approaches enabled a comparative analysis of sequence features across diverse domains of life, thereby forming the basis for our evolutionary investigation.
A significant advancement in the understanding of CRISPR loci occurred in 1995 when a scientist from the University of Italy identified repetitive DNA structures in the genome of an archaeal organism, revealing similarities to those previously described in bacterial genomes 10,11. This observation led to the early hypothesis that such sequences contain foreign DNA fragments and function as part of an adaptive immune system in both bacteria and archaea 12-14. Since then, the CRISPR-Cas system has been increasingly recognized as a powerful defense mechanism and genetic tool. CRISPR loci exhibit a high degree of polymorphism across bacterial strains, including pathogenic species, which has enabled their application in microbial typing and clinical diagnostics. Among the major CRISPR systems, CRISPR-Cas12a is distinguished by its recognition of T-rich PAM sequences, expanding its utility for genome editing in regions inaccessible to CRISPR/Cas9 15-17. The CRISPR/Cas9 system itself originates from a natural bacterial defense mechanism. During viral infection, bacteria incorporate short DNA fragments of the invader into their genome, forming CRISPR arrays. These sequences are then transcribed into crRNAs, which guide the Cas9 endonuclease to target and cleave matching sequences in invading DNA, thereby neutralizing the threat 18-20.
CRISPR-Cas systems are categorized into two major classes, six types, and 33 subtypes. Class 1(types I, III, IV) includes systems with multi-protein effector complexes, while Class 2 (types II, V, VI) contains single multidomain effector proteins such as Cas9, which is characteristic of type II systems found predominantly in bacteria 21-24. However, the classification of CRISPR-Cas systems is complicated by the emergence of hybrid loci formed through extensive recombination events. These hybrid systems often defy standard classification despite containing canonical Cas genes. Furthermore, multiple CRISPR-Cas systems may coexist within a single genome, and even strains of the same species may carry distinct system types 25.
Many CRISPR-Cas loci are embedded in genomic islands that also encode mobile genetic elements such as transposases, toxin-antitoxin modules, and various defense-related genes. The distribution of CRISPR types is non-uniform among microorganisms: type II systems have been detected only in bacteria, while type III systems are more common in archaea. This pattern is consistent with earlier findings that suggest CRISPR systems are generally more prevalent in archaeal lineages than in bacterial ones 26-35.
Despite the promise of CRISPR/Cas9 for antimicrobial applications, one of the significant limitations is the challenge of effective delivery into bacterial cells. While plasmid-based electroporation remains the dominant method for introducing CRISPR components in vitro, it is often impractical for in vivo use 5,36.
Currently, three primary formats are employed for CRISPR delivery: (1) plasmids encoding both Cas9 and sgRNA, offering a stable DNA-based platform but requiring nuclear entry (Figure 2); (2) RNA-based approaches using sgRNA and mRNA encoding Cas9, which are safer and do not integrate into the host genome; and (3) preassembled Cas9 protein-sgRNA complexes (RNA-AP format), which minimize integration risks and can be directly functional (Table 1).
Although the CRISPR-Cas system has been extensively studied for its role in microbial immunity and gene editing, its potential evolutionary or functional relationship with eukaryotic small RNAs such as microRNAs (miRNAs) remains largely unstudied. Therefore, this study aims to investigate the sequence similarities between CRISPR loci in archaea and bacteria and eukaryotic miRNAs, with the objective to uncover potential evolutionary links or functional convergence between these RNA-based regulatory systems.
Materials and Methods :
CRISPR/Cas++ server: In the present study, the CRISPR/Cas++ tool (version 1.1.2, 2021, I2BC) was utilized to extract repetitive sequences from prokaryotic genomes. From a pool of 100 randomly selected prokaryotic species, sequences corresponding to 20 genes and 20 species of bacteria, as well as 20 genera and 20 types of archaea, were retrieved using the miRBase database. These repetitive sequences were subsequently compared with eukaryotic microRNAs obtained from the same database to identify potential sequence similarities. The analysis revealed that only three eukaryotic microRNAs exhibited similarity to the prokaryotic sequences_ specifically, one gene from a bacterial species and one gene from two archaeal species, suggesting a potential role in inter-domain biological communication. It is also noteworthy that some bacterial strains lacked identifiable CRISPR or Cas elements, as reported by the CRISPR/Cas++ tool (Table 2).
miRBase server: The key features of miRBase are designed to achieve five main objectives: (1) To establish a standardized naming system for microRNAs; (2) To collect and curate all known microRNA sequences; (3) To provide both human-and machine-readable information for each microRNA; (4) To offer basic supporting evidence for each microRNA (5) and to integrate and provide information regarding microRNA target interactions.
The latest published version (version 22) of miRBase contains information on 38,589 microRNA precursors and 48,860 mature microRNA sequences from 271 different species. Additionally, the database includes 1,493 small RNA sequencing datasets, encompassing more than 5.5 billion reads mapped to microRNAs.
NCBI server: The National Center for Biotechnology Information (NCBI), part of the National Library of Medicine (NLM), has implemented initial updates to several of its services to support NIH-funded researchers and institutions in complying with the 2024 NIH Public Access Policy, which took effect on July 1, 2025. As part of this effort, and using tools available on the NCBI platform (version 3.42.0), the similarity index and degree of sequence conservation among repetitive elements from two archaeal species and their related micro-organisms were analyzed. The comparative analysis results are shown in (Figure 3). Through the use of the NCBI server and by entering the target gene, information regarding its function and the organisms in which it is found was obtained.
HUGO gene server: HUGOgene is a widely used platform for examining and analyzing genetic variants, genome sequences, and DNA sequence data. Key features of this server include: (1) Genetic data analysis; (2) Variant investigation; (3) Genetic disease diagnosis; (4) Gene and protein identification and characterization.
The HUGO Gene Nomenclature Committee (HGNC) is responsible for assigning a unique and ideally meaningful name and symbol to every human gene. The HGNC database currently contains over 24,000 publicly accessible records, each providing approved gene nomenclature and associated gene information. Recently, the HGNC database was relocated to the European Bioinformatics Institute (EBI). It now offers direct access to various integrated resources, including the searchable HGNC database, the HCOP orthology prediction tool, and manually curated gene family web pages.
In this study, the HUGOgene was used to query a variety of genes and vary their scientifically accepted and officially approved names.
GeneCards server: For over two decades, GeneCards has served as a comprehensive gene-centric database, automatically mining and integrating information from a wide range of data sources. This process results in a web-based card for each of the tens of thousands of human genes. Developed and maintained by the Department of Molecular Genetics at the Weizmann Institute of Science, GeneCards was established in 1997 with the goal of unifying fragmented genetic information from various specialized databases into a coherent and accessible resource. In the present study, utilizing GeneCards at the RNA center, key information regarding the target gene was obtained, including its chromosomal location, exon-intron structure, and potential associations with human diseases.
Results :
In this study, the findings focus on three prokaryotic species: one bacterial and two archaeal. Initially, the repetitive sequences of these species were identified using the CRISPR/Cas system and then compared with repetitive sequences from human microRNAs obtained from the miRBase database. The comparison aimed to identify similarities between bacterial and human microRNAs. Table 3 presents detailed information on several human microRNAs identified during this process. The first column lists the microRNAs, each labeled with the prefix-hsa (Human sapiens), while the second column provides their unique accession numbers from the miRBase database, shown as MIMAT codes, which are essential for accurate identification.
Yersinia ruckeri (Y. ruckeri): Initially, Yersinia bacteria were submitted to the CRISPR/Cas++ server, where their repetitive sequence were obtained. Following this, human microRNAs similar to the identified sequences were determined using the miRBase server (Figure 4). On the other hand, the multi-gene card server includes genes such as FOXO1, PAX7, PTEN, and DOCK3, which are involved in these microRNAs. Additionally, several diseases are associated with these microRNAs, including lung cancer, muscular dystrophy, and Duchenne muscular dystrophy (Table 4 and Figure 5). According to NCBI, sequence IDs are mentioned in supplementary figures S1-S4.
The FOXO1 gene belongs to the forkhead family of transcription factors, characterized by a conserved forkhead domain. Although its exact function is not yet fully understood, FOXO1 is believed to play a role in the regulation of cellular growth and differentiation. Genetic alterations in this gene have been associated with the development of alveolar rhabdomyosarcoma. In this study, the gene of interest was compared with its mutated variant using the BLAST tool, and the results are illustrated in figure S1. The findings suggest that FOXO1 may be involved in mitogen-induced growth and differentiation. To further investigate, the gene's FASTA sequence was retrieved and subjected to BLAST analysis, which revealed several mutations linked to human diseases.
A comparative analysis between the wild-type and mutated forms of the gene indicated that approximately 20% of disease-associated mutations in humans are localized within the FOXO1 gene. These pathogenic mutations were identified using the BLAST+ (version 2.11.0) server, specifically through BLASTn, which was employed throughout this study for similarity searches using the gene's FASTA format.
The PAX gene belongs to the transcription factor family and is typically characterized by the presence of a paired box domain, an octapeptide, and a homeodomain. Members of this gene family are known to play critical roles in embryonic development and cancer progression. Although the precise biological function of PAX7 is not yet fully understood, it is hypothesized to function as a tumor suppressor, particularly due to its fusion with members of the alveolar forkhead family. PAX7 is believed to contribute to both fetal growth and development and tumorigenesis.
Multiple mutations have been identified within the PAX7 gene, many of which are associated with human diseases, possibly due to sequence similarities with other genes. According to data retrieved from the NCBI database, comparative analysis between the standard and mutated forms of the gene revealed that approximately 40-60% of the mutations occurring in PAX7 are pathogenic.
These mutations were identified using the BLAST+ (version 2.11.0) server, with BLASTn format, and the results are presented in figure S2.
PTEN has been widely recognized as a tumor suppressor gene in various types of cancers, primarily due to the function of its encoded protein, which acts as a phosphatidylinositol-4,5-bisphosphate 3-phosphatase. This protein contains a tensin-like domain and a catalytic domain that resembles those found in dual-specificity protein phosphatases. Unlike many protein tyrosine phosphatases, the PTEN protein preferentially dephosphorylates phosphoinositide substrates. By reducing intracellular levels of phosphatidylinositol-4,5-bisphosphate, it negatively regulates key signaling pathways, thereby exerting its tumor suppressor function (Figure S3).
To identify pathogenic mutations, the BLAST server (version 2.11.0+) was used. Throughout this study, BLASTn was employed for all sequence comparisons. Similarity searches were conducted using the FASTA format of the PTEN gene.
DOCK3 is selectively expressed in the central nervous system and encodes a member of the Guanine nucleotide Exchange Factor (GEF) family. The encoded protein, also known as cytokine 3, functions both as a regulator of cell adhesion and as a presenilin-binding protein. It facilitates axonal growth within the central nervous system by promoting membrane trafficking and activating G proteins (Figure S4).
Pathogenic mutations in this gene were identified using the BLAST server (version 2.11.0+). Throughout this study, BLASTn was utilized for all sequence analyses. Similarity searches were conducted using the FASTA format of the DOCK3 gene.
Acidianus ambivalens (A. ambivalens): In the first instance, by inputting A. ambivalens into the CRISPR/Cas++ server, its repeated sequence was obtained. After obtaining the repeated sequence of A. ambivalens, using the miRBase server, human microRNAs similar to the mentioned sequences were identified.
According to the multi-gene card server, the GRCH38 and CHM13 genes are involved in these microRNAs, and four types of diseases can also be associated with these microRNAs. Such as adenocarcinoma of lung, thyroid neoplasms, stomach neoplasms and, uterine cervical neoplasms, which are related to these microRNAs, were identified (Table 5). According to NCBI, sequence IDs are listed in supplementary figures S5-S7.
The CFH gene, as annotated in the CHM13 genome, encodes a secreted protein that belongs to the complement factor H protein family. This protein interacts with Pseudomonas aeruginosa elongation factor Thf, in conjunction with plasminogen, which subsequently undergoes proteolytic activation. It has been proposed that Tuf functions as a virulence factor by recruiting host proteins to the bacterial surface, thereby modulating complement activity and facilitating tissue invasion. Mutations in the CFH gene have been associated with an increased risk of atypical Hemolytic-Uremic Syndrome (aHUS) [RefSeq, Oct 2009].
To identify pathogenic mutations, the BLAST+ v2.11.0 server was used, with BLASTn employed for the analysis. Similarity searches were conducted using the FASTA format of the CFH gene sequence. The results are illustrated in figure S6.
The TP53 gene, as annotated in the GRCh/hg38 reference genome, encodes a tumor suppressor protein that contains transcriptional activation, DNA-binding, and oligomerization domains. This protein plays a critical role in responding to various cellular stress signals by regulating the expression of target genes involved in cycle arrest, apoptosis, senescence, DNA repair, and metabolic processes.
Mutations in TP53 are associated with numerous human cancers, including hereditary cancer syndromes such as Li-Fraumeni syndrome. Alternative splicing events and the use of alternate promoters result in multiple transcript variants and protein isoforms. Furthermore, additional isoforms may arise from the use of alternative translation initiation codons within the same transcript variants (PMIDs: 12032546, 20937277) [RefSeq, Dec 2016].
This gene has also been reported to be associated with the microRNA as mentioned above. To identify potential pathogenic mutations, the BLAST+ v2.11.0 server was used, employing the BLASTn algorithm throughout the study. Sequence similarity searches were performed using the FASTA format of the TP53 gene. The results are presented in figure S7.
Acidianus S.P: Initially, the sequence of archaea was obtained by inputting it into the CRISPR/Cas++ server. These microRNAs are linked to several human diseases, including rheumatoid arthritis, Alzheimer's disease, thyroid cancer, and carcinoma. Using the GeneCards server, the genes involved in these microRNAs and the diseases associated with them were identified. According to the results, the server highlighted the following genes: Irak2, NOS2, Hipk3, Numb, and STAT1. Further details are provided below.
Psoriatic arthritis is a disease characterized by joint inflammation (arthritis) that is often associated with a skin condition called psoriasis. It is a chronic inflammatory disease marked by patches of red, irritated skin, usually covered with scaly, white crusts. People with this condition may also experience changes in their fingernails and toenails, such as pitting, thickening, crumbling, or separation from the nail bed.
Rheumatoid arthritis is an autoimmune disorder characterized by pain, swelling, stiffness, and joint damage. While it can affect any joint, it most commonly impacts the wrists and fingers. This condition is more prevalent in women than men and typically onset in middle age. Rheumatoid arthritis development is influenced by a combination of genetic factors, environmental triggers, and hormonal changes. Treatment strategies include pharmacological interventions, lifestyle modifications, and surgical options, all of which aim to alleviate symptoms, reduce pain and swelling, and slow disease progression.
Non-medullary thyroid cancer refers to malignancies originating from follicular cells, which represent more than 95% of all thyroid cancer cases. Cancers arising from parafollicular cells are comparatively rare. Hepatocellular carcinoma is the most prevalent form of primary malignant liver tumor, ranking as the fifth most common cancer globally and the third leading cause of cancer-related mortality. The primary risk factors for hepatocellular carcinoma include chronic infection with hepatitis B or C viruses, prolonged exposure to aflatoxin-contaminated food, and excessive alcohol consumption. Hepatoblastoma, which constitutes 1-2% of pediatric malignant neoplasms, predominantly affects children under the age of three (Table 6) (Figures 6 and 7). According to NCBI, sequence IDs were mentioned in supplementary figures S8-S13.
STAT1 functions as a key activator within various signaling pathways and is encoded by the STAT1 gene. This protein is activated by interferons α and γ, as well as by Epidermal Growth Factor (EGF). STAT1 plays a crucial role in the immune response against a range of pathogens, including viruses, fungi, and mycobacteria, and is also involved in signaling responses to cytokines and growth factors. Upon activation, STAT1 undergoes phosphorylation, resulting in the formation of homo-or heterodimers, often involving other STAT family members and associated kinases (Figure S8).
To identify pathogenic mutations, the BLAST+ v2.11.0 server was used, with the BLASTn algorithm applied throughout the study. Similarity searches were conducted using the FASTA sequence format of the STAT1 gene.
The protein Irak2 encoded by this gene belongs to the STAT1 protein family, which, in response to cytokines and growth factors, undergoes phosphorylation by receptor-associated kinases. These phosphorylated proteins subsequently form stable homo- or heterodimers, which are translocated to the cell nucleus, where they function as transcriptional activators. The encoded protein can be activated by a variety of ligands, including interferons alpha and gamma, interleukin-6, EGF, and Platelet-Derived Growth Factor (PDGF). This protein plays a critical role in regulating the expression of genes essential for cell survival in response to various cellular stimuli and pathogens, and it is vital in mediating immune responses to infections caused by pathogens, viruses, and mycobacteria. Mutations in this gene have been linked to immunodeficiency (Figure S9). To identify pathogenic mutations, the BLAST+ v2.11.0 server was used, with the BLASTn algorithm applied throughout the study. Similarity searches were conducted using the FASTA sequence format of the Irak2 gene.
NOTCH1 encodes a protein that is a member of a family of membrane-bound proteins, characterized by structural features such as an extracellular domain composed of multiple EGF repeats and an intracellular domain containing several distinct domain types. This gene is a key component of an evolutionarily conserved intercellular signaling pathway that governs the interactions between neighboring cells (Figure S10). Pathogenic mutations were identified using the BLAST server (BLAST+ version 2.11.0). All analyses in this study were conducted using the BLASTn algorithm. Similarity searches were performed based on the FASTA format of the target gene.
Nitric oxide, also known as NOS2, is an active free radical that serves as a biological mediator in various physiological processes, including neurotransmission, antimicrobial defense, and antitumor activity. This gene encodes a nitric oxide synthase enzyme that is primarily expressed in the liver and is upregulated by lipopolysaccharides and specific cytokines (Figure S11). Pathogenic mutations were identified using the BLAST server (BLAST+ version 2.11.0). The BLASTn algorithm was employed throughout the study. Sequence similarity searches were conducted using the FASTA format of the gene.
The Hipk3 gene activates serine/threonine kinase activity and negatively regulates the kinase. By analyzing the sequence of the healthy gene and comparing it with the defective one, it was determined that nearly 30% of the mutations occurred in the faulty gene (Figure S12). Pathogenic mutations were identified through analyses performed on the BLAST server (BLAST+ v2.11.0). All similarity searches in this study utilized the BLASTn algorithm, with queries conducted using the FASTA format of the target gene.
The final gene, Numb, is involved in the regulation of cell fate determination during development. The protein encoded by this gene undergoes degradation in a proteasome-dependent manner, facilitated by a membrane-bound protein (Figure S13). Pathogenic mutations were identified using the BLAST server (BLAST+ v2.11.0), with all analyses conducted using the BLASTn algorithm. Sequence similarity searches were performed based on the gene's FASTA-formatted sequence.
Finally, the E-values (set to 1e-5), identity percentages, and query coverage provided by the respective servers support the purported sequence similarities.
Discussion :
About ten years ago, the emergence of CRISPR marked a significant turning point in the field of genome editing, suggesting a more precise and cost-effective alternative to earlier methods. By studying and engineering Cas enzymes derived from different bacterial species, the technology rapidly advanced, gaining impressive versatility in a relatively short time. This quick evolution has accelerated scientific research, allowing researchers worldwide to investigate a wide range of biological processes with greater accuracy and depth.
Despite its many benefits, CRISPR also presents several challenges, including variable editing efficiency, reliance on clonal selection, and the risk of off-target effects. These issues are particularly relevant in cancer research, where tumor heterogeneity complicates the isolation of CRISPR-modified clones. Cells with different drug responses or stages of differentiation may be lost during selection, making it challenging to generate models that accurately reflect the complexity of the original tumor.
Even so, CRISPR has already established itself as an invaluable tool for studying the molecular basis of cancer and for dissecting the interactions between specific pathways and genes. So, it is expected to play an increasingly important role in both basic and translational research. Further studies will likely refine CRISPR's utility in probing in vivo biological mechanisms using more advanced, clinically relevant models. Additionally, its applications are expected to expand in areas such as adoptive cell therapies, which are becoming central to both cancer treatment and the management of degenerative diseases 35.
It is critical to emphasize that the findings of this study are based on computational analyses using public databases and sequence alignment tools. Although these in silico approaches can provide valuable preliminary insights into the possible similarities between CRISPR loci and human microRNAs, they are no substitute for laboratory confirmation. Experimental validation or functional tests are necessary to confirm the biological significance and functional implications of these observed similarities. Without such validation, the results should be interpreted with caution, as they may be affected by the quality of the databases or the algorithmic limitations of BLAST and other bioinformatics tools. Therefore, future studies should incorporate laboratory-based experiments to substantiate the computational predictions of this study and increase the robustness of the results.
Given that some of these sequences align with human microRNAs and are associated with mutations in disease-related genes, this area of research holds considerable promise. It raises crucial questions about cross-domain interactions and warrants further investigations to understand its implications for human health better.
Conclusion :
Given the complexity and highly dynamic evolution of CRISPR/Cas9 systems, attempting to classify them based on a single criterion—such as the phylogenetic analysis of Cas1—would be inadequate and potentially misleading. A comprehensive classification requires consideration of multiple factors to reflect the diversity and functional variability of these systems accurately. In this study, the repetitive sequence of Y. ruckeri and two different species of archaebacterium, Acidianus, were obtained using the CRISPR/Cas9 system. Similar microRNAs from humans and animals, as identified on the miRBase site, were then compared with these sequences, and the degree of similarity with microRNAs was determined. In the continuation of the research, genes involved in bacteria and archaea were obtained using NCBI servers and GeneCards. Moreover, the diseases associated with human microRNAs similar to the bacterial and archaeal repetitive sequences were identified using the RNAcenter section of the GeneCards server 36. Table 8 summarizes the relevant results.
Acknowledgement :
The authors thank the Department of Biotechnology, Faculty of Advanced Sciences and Technologies, University of Isfahan, for supporting this study.
This work should be attributed to the Faculty of Biotechnology, Isfahan University, Isfahan, Iran.
Conflict of Interest :
The authors declared no conflict of interest related to this article.
Funding: This work should be attributed to the Faculty of Biotechnology, Isfahan University, Isfahan, Iran.
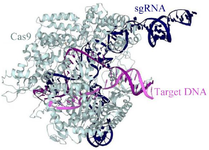
Figure 1. Crystallographic studies have shown that Cas9 consists of two main lobes: a recognition lobe and a nuclease lobe. The binding of guide RNA to target DNA induces the formation of a positively charged groove at the interface of these lobes 10.
|
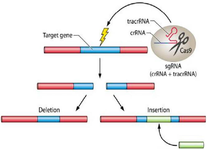
Figure 2. The CRISPR system utilizes a single guide RNA (sgRNA) composed of a target-matching sequence and an activating region that recruits Cas9. This complex precisely induces double-strand breaks at specific genomic sites, leading to gene disruption. Owing to its high accuracy and efficiency, CRISPR is widely adopted in molecular biology and genetic engineering.
|
Figure 3. Obtaining the similarities of repetitive sequences in prokaryotes with other organisms, as well as determining the index.
|

Figure 4. Human miRNAs similar to the Yersinia ruckeri bacterial sequence were identified using the miRBase server.
|
Figure 5. Involves retrieving microRNA accession numbers and identifying related human diseases.
|
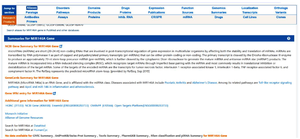
Figure 6. A summary of has-miR146a is provided, along with supporting evidence and its corresponding accession number.
|
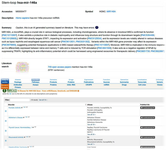
Figure 7. Several human diseases are associated with this microRNA, according to data from the GeneCards database.
|

Figure S1. BLASTn-based graphical comparison of FOXO1 gene sequences showing 30% mutation relative to the reference FASTA.
|

Figure S2. Comparative BLASTn analysis of PAX7 gene sequences revealed~30% mutation by aligning the reference FASTA sequence with defective variants, highlighting key sequence similarities and mutation sites.
|
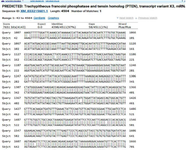
Figure S3. BLASTn-based graphical comparison of PTEN gene sequences showed~30% mutation by aligning defective sequences with the reference FASTA, revealing conserved regions and mutation sites.
|

Figure S4. By acquiring the sequence of the healthy gene and comparing it to the defective gene, it was found that 10% to 20% of mutations were present in the gene.
|
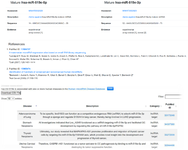
Figure S5. Some human diseases are associated with miR-519e, and their corresponding accession numbers were also retrieved.
|

Figure S6. Approximately 30% of the gene was found to be mutated by comparing the defective sequence with the reference gene in FASTA format.
|

Figure S7. Using BLASTn alignment, approximately 10% mutation was identified in the defective sequence of the TP53 tumor suppressor gene compared to the wild-type in the GRCh38 reference genome.
|

Figure S8. By comparing the FASTA format of the reference gene with the gene, it was observed that the gene has a 10% mutation responsible for the disease.
|
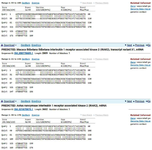
Figure S9. Analysis of the Irak2 gene revealed 60-70% mutation frequency compared to the healthy reference, based on NCBI data. These mutations, linked to immunodeficiency, affect the interleukin-1 receptor-associated kinase 2, a key regulator of IL-1-induced NF- κB signaling.
|

Figure S10. FASTA comparison shows ~40% mutation presence in NOTCH1, associated with aortic valve disease and T-cell acute lymphoblastic leukemia.
|

Figure S11. Detailed molecular analysis revealed that 16% of mutations are localized within the defective allele, providing further validation of this observation.
|
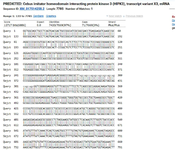
Figure S12. Comparative analysis of the FASTA sequences of the healthy and mutated Hipk3 genes revealed that approximately 20% of the total observed variations are found in the mutated sequence.
|

Figure S13. FASTA comparison demonstrates~30% of mutations in the defective gene, with additional Numb gene mutations identified via BLAST analysis on the NCBI server.
|

Table 1. The three types of transmission for the CRISPR/Cas9 system and their characteristics 5
|

Table 2. Sequences related to bacteria and archaea were obtained using the CRISPR-Cas++ server
|

Table 3. microRNAs accession numbers
|

Table 4. Overview of Yersinia ruckeri (Y. ruckeri) species, their similarity to human genes, and related diseases
|

Table 5. Summary of Acidianus ambivalens (A. ambivalens), similarity to human microorganisms, genes involved in this microorganism, and associated diseases
|

Table 6. A summary of archaea repetitive sequences, their similarity to human microRNAs, the genes associated with these microRNAs, and the related diseases is provided
|
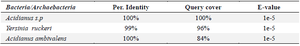
Table 7. BLAST-based analysis showing sequence identity, coverage, and E-values for bacterial and archaeal species aligned with the target gene
|
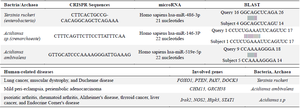
Table 8. Summary of the examined bacterial names, their repetitive sequences, corresponding microRNA sequences, and their similarity using CRISPR/Cas++, miRBase, and BLAST sites
|
|