Exploring the Role of hsa_circ_0052112 as a Potential Biomarker in Breast Cancer: Insights from Experimental and In Silico Analyses
-
Alizadeh, Mahdi
-
Department of Medical Genetics, Institute of Medical Biotechnology, National Institute of Genetic Engineering and Biotechnology (NIGEB), Tehran, Iran
-
 Salimi, Mahdieh
Department of Medical Genetics, Institute of Medical Biotechnology, National Institute of Genetic Engineering and Biotechnology (NIGEB), Tehran, Iran, Tel: +98 9123341722, 44787382; Fax: +98 21 44787395; E-mail: salimi@nigeb.ac.ir
Salimi, Mahdieh
Department of Medical Genetics, Institute of Medical Biotechnology, National Institute of Genetic Engineering and Biotechnology (NIGEB), Tehran, Iran, Tel: +98 9123341722, 44787382; Fax: +98 21 44787395; E-mail: salimi@nigeb.ac.ir
Abstract: Background: Circular RNAs (circRNAs) are important in tumorigenesis and cancer progression, highlighting their potential as biomarkers for diagnosis, prognosis, and treatment monitoring.
Methods: This study consists of experimental and in silico phases. In the experimental phase, the expression of hsa_circ_0052112 in tumor and blood samples from 40 breast cancer women was analyzed, compared to the control group using Sybr Green real-time RT-PCR followed by total RNA extraction and cDNA synthesis. Statistical analysis was performed using the beta-actin gene as a normalizer, compared to the normal control group as a fold change. In the in silico phase, interactions among circRNA, RNA-Binding Proteins (RBPs), and microRNAs (miRNAs) were investigated using Interactome Database. The miRcancer database was utilized to assess breast cancer-related miRNAs linked to hsa_circ_0052112. Target miRNAs were identified with TargetScan and filtered for relevance through DisGeNET. K-means clustering grouped genes by expression patterns, visualized in Cytoscape to illustrate circRNA-miRNA-mRNA relationships. Hub genes underwent pathway enrichment analysis using Reactome database to determine their functional significance.
Results: Data revealed a significant increase in hsa_circ_0052112 expression in both blood and tumour of breast cancer patients. This increase was especially pronounced in patients with estrogen, progesterone, and HER2 receptor positivity, as well as in advanced disease stages with lymph node involvement. Enrichment analysis of hub genes indicates their role in the PI3K/AKT signaling pathway.
Conclusion: hsa_circ_0052112 shows promise as a multifaceted biomarker for breast cancer, enhancing diagnosis and prognosis; while supporting personalized treatment strategies. Further clinical validation is necessary to confirm its utility.
Introduction :
Heritable and dynamic changes occurring to the genome independently of DNA sequence are known as "epigenetics". Cancer was the first disease reported as an epigenetic changes-related disease 1. The activation of oncogenes and/or the suppression of tumor suppressor genes are critical events always accompanied by epigenetic changes. These epigenetic changes include DNA methylation, micro-RNA and long non-coding RNA expression, and histone post-translational modifications 2,3. Breast cancer development is a multistep and complex process that includes the synergistic crosstalk between epigenetic and genetic alterations, which are influenced by an abundance of external and internal factors. Such factors include but are not limited to the cell’s intrinsic nutrient supply, microenvironment, cellular stress, and external environmental exposures to endocrine disruptors or carcinogenic agents. The epigenetic changes in breast cancer development and progression are influenced by critical genes involved in apoptosis, proliferation, cell motility, invasion, and other processes 4,5. Modification of epigenetic changes has been reported as a promising therapeutic strategy in breast cancer. Breast cancer is a heterogeneous type of malignancy that results from unique molecular changes in breast tissue 6.
Circular RNAs (circRNAs) are stable and evolutionarily conserved RNA regulators that can act as microRNA (miRNA) sponges, regulate alternative splicing mechanisms, and have an influential role in the expression of the encoded genes 7. CircRNAs have been shown to play essential roles in the cell and are one of the most crucial research focuses, particularly in cancer 8. Due to their significant roles in critical genetic pathways, particularly in oncology, two distinct therapeutic potentials have been identified for circRNAs: inhibiting carcinogenic circRNAs or restoring circRNAs that possess tumor suppressor functions 9. Since molecular phenotypes do not operate in isolation, but their interactions collectively carry out biological processes, it is essential to model these processes in living cells using networks 10,11. It is anticipated that identifying circRNA-miRNA-mRNA connections will be crucial for explaining the molecular processes underlying numerous diseases, detecting biomarkers for early diagnosis, and expanding therapeutic options. A single miRNA can target hundreds of genes, while a single circRNA can act as a sponge for tens of miRNAs. Using bioinformatics data to simplify the circRNA-miRNA-mRNA interactions may shed light on in vitro and in vivo investigations 12.
In this study, focus was on hsa_circ_0052112, a circular RNA derived from the ZNF83 gene. This selection was based on circRNA expression profile analyses in breast cancer cells, which were conducted using a microarray assay in 2018. The expression levels of hsa_circ_0052112 in MCF-7 and MDA-MB-231 cells were measured using RT-qPCR, along with evaluations of the migration and invasion capabilities of breast cancer cells. The findings indicated that the expression of hsa_circ_0052112 was significantly higher in MDA-MB-231 cells compared to MCF-7 cells. Additionally, the overexpression of hsa_circ_0052112 promoted cell migration and invasion in breast cancer, whereas its downregulation suppressed these processes 13. As of now, no additional studies have been published on hsa_circ_0052112. Considering its significant implications and established relevance to breast cancer, researchers set out to build upon this research by investigating the expression of hsa_circ_0052112 in tumor and blood samples from breast cancer patients in comparison to normal controls. Additionally, the aim was to examine its expression across various clinicopathological groups for the first time.
The present research was designed in experimental and in silico phases. In the experimental phase, the expression of hsa_circ_0052112 in the tumor tissue and blood of breast cancer patients was investigated in comparison with the normal control group, and its association with the clinical characteristics of the disease was studied. In the in silico phase, the interaction of hsa_circ_0052112 with miRNA, RBPs, and target genes was investigated. Breast cancer remains a leading cause of cancer-related morbidity and mortality among women worldwide, necessitating the identification of novel biomarkers for improved diagnosis, prognosis, and treatment strategies. Circular RNAs have emerged as significant regulatory molecules in various biological processes, including tumorigenesis and cancer progression, highlighting their potential utility in oncology 14. This study specifically investigates the expression of hsa_circ_0052112 in breast cancer, aiming to elucidate its role as a biomarker. By analyzing hsa_circ_0052112 levels in tumor and blood samples from breast cancer patients and correlating these findings with clinical characteristics, researchers seeked to establish a direct link between circRNA expression and breast cancer phenotypes. Furthermore, examination of the interactions between hsa_circ_0052112, RNA-binding proteins (RBPs), and microRNAs (miRNAs) aims to provide insights into the molecular mechanisms underlying its involvement in breast cancer. Ultimately, this research endeavors to contribute to the development of personalized treatment strategies by establishing hsa_circ_0052112 as a multifaceted biomarker with significant clinical relevance.
Materials and Methods :
Experimental phase: Sampling: 40 blood and breast tissue (Tumor and Normal adjacent) samples were collected from breast cancer patients at Imam Khomeini Hospital in Tehran, Iran. A control blood group consisting of 40 samples was also collected from non-affected individuals. The inclusion criteria for the normal control group samples were female participants within the age range of the patients, devoid of any history of breast disease or malignancy in both themselves and their first-degree relatives. The inclusion criteria for the test group were patients who were diagnosed with ductal carcinoma of the breast and had not initiated any treatment at the time of sampling. The demographic and clinical data of the patients and controls have been included in table 1. Ethical considerations and informed consent paperwork had been acquired for this study (IR.MODARES.REC.1398.148).
RNA extraction: Peripheral blood samples (3-4 ml) were collected into EDTA tubes for plasma preparation. The tubes were centrifuged at 1000 g for 15 min within 30 min of collection. Subsequently, 1 ml of plasma was transferred to a 1.5‐ml Eppendorf tube and centrifuged at 11,000 g for 10 min to remove any remaining cellular debris. Total RNA was extracted from plasma and breast tissue samples using the RNX-Plus solution (CinnaClone-Iran) following a multi-step protocol. The process began with the addition of RNX-Plus to lyse cells and release RNA, followed by the introduction of chloroform for phase separation, which involved vigorous shaking and centrifugation to isolate the RNA-containing aqueous phase. RNA was then precipitated by adding isopropanol or ethanol and incubating the mixture at -20°C. After precipitation, the RNA pellet was washed with 70% ethanol to eliminate impurities and air-dried, before being resuspended in RNase-free water or buffer. The concentration and purity of the extracted RNA were assessed using a NanoDrop ND-ONE spectrophotometer (Thermo-USA), measuring absorbance at 260 nm and calculating the 260/280 ratio to check for protein contamination.
cDNA synthesis: For cDNA synthesis, the YT4500 cDNA Synthesis Kit (Yekta Tajhiz Azma, Iran) was utilized. The process began with the preparation of a reaction mixture comprising extracted RNA, random hexamer primers from the kit, reverse transcriptase, dNTPs, and buffer components. This mixture was then incubated according to the manufacturer's protocol, which involved an initial primer annealing at 25°C for 10 min, followed by reverse transcription at 42°C for 60 min, and enzyme inactivation at 70°C for 5 min. The synthesized cDNA was subsequently stored at -20°C for future use in quantitative RT-PCR.
SYBR Green Quantitative real-time reverse transcription PCR: To quantify the expression levels of circRNA (Hsa_circ_0052112) and the internal control gene β-actin, SYBR Green real-time RT-PCR was utilized, allowing for the amplification and detection of specific DNA sequences in real-time. β-actin was selected due to its stable expression across various cell types and experimental conditions, demonstrated minimal variability in preliminary assessments compared to other housekeeping genes, and extensive literature validation, thereby ensuring our findings' robustness and reliability. Each PCR reaction contained a cDNA template, specific primers for hsa_circ_0052112, forward: AGAGGGCTTTATACAGGGCC; reverse: CCCTGA AAGTCAAGCATCCC, and β-actin (forward: CACCTTCTACAATGAGCTGCGTGTG; reverse: ATAGC ACAGCCTGGATAGCAACGTAC, SYBR Green dye, and PCR buffer. The thermal cycling conditions included an initial denaturation at 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 30 s, annealing at 57°C for 30 s, and extension at 72°C for 30 s. Each reaction incorporated negative controls (lacking template DNA) to check for contamination and technical duplicates to ensure reproducibility. The relative expression of circRNA was normalized to β-actin, with primer efficiency evaluated using LinRegPCR software to confirm approximately 100% efficiency. The comparative quantification of gene expression was conducted using the 2-ΔΔCT method, where ΔCT represents the difference in Cycle threshold (Ct) values between the target gene and β-actin. Expression changes were categorized as upregulated (two-fold or higher), normal (between 0.5 and 2-fold), or downregulated (0.5-fold or lower), ensuring accurate and reliable quantification of gene expression levels in the analyzed samples.
Statistical analyses: The acquired data were analyzed using Graph Pad Prism software version 8.2. Since the data showed a normal distribution, parametric tests were employed for statistical analysis. To compare circRNA expression between two groups and more than two groups, the T-test and Analysis of Variance (ANOVA) test were conducted. The significance level was set at p<0.05. Results were reported as mean±standard deviation. Cohen's d-test was used for effect size calculation.
In silco phase: Investigating RNA regulatory interactions using the interactome database: The Interactome site (https://circinteractome.nia.nih.gov/) was employed as a supply for investigating the interactions between circular RNA (circRNA), RNA-Binding Proteins (RBPs), and microRNAs (miRNAs) in this study. This database comprises experimentally validated interactions between these molecules, enabling researchers to find the convoluted relationships within the RNA regulatory network. By analyzing the interactions on the Interactome site, we gained insights into how circRNAs, RBPs, and miRNAs interact, which may indicate novel regulatory mechanisms and biological functions. This information was significant for realizing gene expression regulation, disease mechanisms, and identifying potential therapeutic targets in the study performed. Table 2 shows the characteristics of circular RNA taken from the interactome site.
Investigating the interaction between RNA-binding proteins and circular RNA in post-transcriptional: Gene Regulation: The correlation between RBPs and circRNAs is especially noteworthy in RNA biology. RBPs can bind to circRNAs, touching their stability, localization, and potential functions. This interaction may result in the formation of circRNA-protein complexes that can serve as regulators of gene expression or signaling pathways. Comprehension of the relationship between RBPs and circRNAs is crucial for clarifying the complex regulatory networks within cells and revealing the roles of circRNAs in various biological processes. RNA-binding protein sites matching flanking regions of circRNA, taken from the interactome site, are shown in table 3.
Exploring the regulatory interplay between microRNAs and circular RNAs using the interactome database: In this study, the interplay between miRNAs and circRNAs engaging the Interactome site to uncover the regulatory mechanisms within the RNA network was explored. miRNAs are small non-coding RNAs that regulate gene expression post-transcriptionally by binding to target mRNAs and regulating their translation or degradation. CircRNAs, alternatively, are manifesting as important gene expression regulators that can segregate miRNAs and modulate their activity. By evaluating the interactions between miRNAs and circRNAs on the Interactome site, researchers intended to reveal new regulatory pathways and potential therapeutic targets. Recognition of how circRNAs impact miRNA function can provide a significant understanding of the complicated dynamics of different RNA molecules and their impact on gene expression. This insight is important for understanding disease mechanisms, identifying biomarkers, and developing targeted therapies for various disorders. Table 4 lists the miRNAs that are targeted by hsa_circ_0052112 taken from the interactome site.
Identification of breast cancer-related miRNAs targeted by circular RNA through enrichment analysis: Among miRNAs targeted by hsa_circ_0052112 (Table 4), enrichment was done using miRcancer database (https://mircancer.ecu.edu/), and miRNAs related to breast cancer were identified.
Visualization of circRNA_miRNA_mRNA relationships in a tripartite manner using cytoscape: In this research study, the target miRNA genes selected from the previous step were identified using the TargetScan database (https://www.targetscan.org/). Subsequently, genes related to breast cancer were filtered using DisGeNET. The breast cancer-related genes were represented as data points in a multidimensional space (string), with each dimension reflecting a specific gene expression value. K-means clustering was employed to group genes with similar expression patterns into clusters. The resulting clusters were visualized as a network with confidence of interaction score of 0.9, where genes were depicted as nodes and relationships between genes were shown as edges based on their expression similarities. This network analysis enabled the exploration of gene interactions and functional relationships within biological systems. Following the creation of the gene network in string format, the data was transferred to Cytoscape for further analysis. Gene hubs were identified, and the circRNA_miRNA_mRNA relationships were visualized using Cytoscape in a tripartite manner.
Exploring Hub genes enrichment by reactome: In this study, hub genes enrichment by the Reactome database (https://reactome.org/) involved the identification of key genes that hold significant importance in biological pathways or processes. Reactome, a curated database of biological pathways, offers detailed information on molecular events and their interrelationships across diverse cellular processes. Through hub gene enrichment analysis using Reactome, researchers can identify genes that serve as central components within specific pathways or networks. This analysis aids in elucidating the functional relevance of these hub genes and their involvement in various biological processes. By leveraging Reactome's extensive pathway data, researchers can gain insights into the functional enrichment of hub genes and their potential roles in complex biological systems.
The flowchart of the research and step-by-step phases of the in silico phase of the study are shown in figure 1.
Results :
hsa_circ_0052112 Expression in Breast Cancer Patients: Tumor tissue and blood plasma analysis: As seen in figure 2, the expression level of hsa_circ_0052112 in the samples of breast cancer patients, both in the tumor tissue and the blood plasma, shows a significant increase compared to the control group (p<0.0001). The mean expression of circular RNA in breast tumors is 7.92±2.85 and in plasma is 3.40±1.68.
Association between circular RNA expression and receptor status in breast cancer patients: Implications for disease progression: In table 5, the average expression of hsa_circ_0052112 in groups with estrogen and progesterone receptors, as well as HER2, demonstrates a significant increase in both tissue and blood plasma levels compared to the corresponding groups lacking these receptors. This finding suggests that hsa_circ_0052112 may play a role in the biology of hormone receptor-positive breast cancer, potentially influencing tumor behavior and response to hormonal therapies.
Moreover, the increase in hsa_circ_0052112 expression is particularly pronounced in patients with metastatic disease and those at advanced stages of cancer. This correlation raises important questions about the potential role of hsa_circ_0052112 as a biomarker for disease progression. Its elevated levels could indicate a more aggressive tumor phenotype, which may be associated with poorer prognoses and the need for more intensive therapeutic strategies.
Additionally, the relationship between hsa_circ_ 0052112 expression and lymph node involvement further emphasize its clinical relevance. In patients with lymph node metastasis, hsa_circ_0052112 expression levels were significantly higher than those without lymph node involvement, suggesting its potential utility as a prognostic marker. This association may provide insights into the metastatic potential of breast tumors and aid in the stratification of patients for tailored therapeutic interventions.
Identification of RNA-binding proteins associated with the flanking regions of hsa_circ_0052112: Using the Interactome database, the RBPs that bind to the flanking region of circular RNA hsa_circ_ 0052112 were identified, which are AGO2, AGO2, EIF4A3, and PTB. Their features and characteristics are listed in table 3.
Identification and characterization of miRNAs targeted by hsa_circ_0052112 with implications for breast cancer: Using the TargetScan database, miRNAs targeted by hsa_circ_0052112 were identified, which consists of 7 microRNAs named hsa-miR-142-5p, hsa-miR-146b-3p, hsa-miR-194, hsa-miR-324-5p, hsa-miR-515-5p, hsa-miR-568, and hsa-miR-924. Their characteristics are listed in table 4. Among miRNAs targeted by circular RNA, enrichment was done using miRcancer database and hsa-miR-194 related to breast cancer, were identified.
Tripartite visualization of circRNA-miRNA-mRNA interactions in breast cancer network: Using TargetScan, the genes targeted by hsa-miR-194 were identified. Among them, genes related to breast cancer were filtered using DisGeNET. In figure 3, the interaction network of these genes is depicted, as can be seen, genes MAPK1, STAT1, TRAF6, and ERRB4 were identified as hub genes. A hub gene is a gene that plays a central role in a biological network, often exhibiting a high degree of connectivity with other genes or proteins. As shown in figure 4, the circRNA_miRNA_mRNA relationships were visualized using Cytoscape in a tripartite manner.
Enrichment analysis of hub genes and their role in PI3K/AKT signaling pathways: In this study, hub genes (MAPK1, STAT1, TRAF6, and ERRB4) enrichment by the Reactome database was done and their significant importance in biological pathways or processes was elucidated and summarized in figure 5. As seen in figure 5, the two important biological processes that statistically showed the highest correlation with these genes are PI5P, PP2A, and IER3 regulate PI3K/AKT signaling R-HSA-6811558 and negative regulation of PI3K/AKT network R-HSA-199418.
Discussion :
circRNAs are stable noncoding RNAs that regulate gene expression in various tissues, including body fluids. This makes them a critical and exciting point of novel research into the pathogenesis of numerous diseases 15. A convergence of data supports that circRNAs are involved in cancer, making them novel biomarkers or potential therapeutic targets 13.
In a study published in 2018, circRNA microarrays analysis indicated elevated hsa_circ_0052112 expression in invasive breast cancer cell lines, further, the expression of hsa_circ_0052112 in MCF-7 cells and MDA-MB-231 cells was verified by RT-qPCR assay 13. The expression of this circRNA in breast cancer patients has not been reported.
The present study investigates the relationship between hsa_circ_0052112 expression and clinical parameters in breast cancer, highlighting its potential as a biomarker. Elevated levels of hsa_circ_0052112 were found in tumor tissues and plasma of breast cancer patients, particularly in advanced stages and those with lymph node involvement, suggesting its role in disease progression and tumor aggressiveness. The correlation between hsa_circ_0052112 expression and hormone receptor status indicate its utility not only as a marker for breast cancer presence, but also in understanding tumor behavior, which may inform treatment responses.
The non-invasive measurement of hsa_circ_ 0052112 in plasma offers a promising approach for monitoring disease progression and treatment efficacy, potentially allowing for its incorporation into routine clinical assessments. Should future studies confirm its prognostic value, hsa_circ_0052112 could aid in patient stratification, guiding personalized treatment strategies based on expression levels.
The traditional markers ER, PR, and HER2 have been extensively validated in clinical settings and play crucial roles in determining the therapeutic approach for breast cancer patients. However, these biomarkers often exhibit variability in expression and can be influenced by tumor heterogeneity, potentially limiting their reliability as sole indicators of disease status 16. In contrast, hsa_circ_0052112 demonstrates a unique profile characterized by its circular RNA structure, which confers increased stability in circulation compared to linear RNA species. This stability is particularly advantageous for its potential application in liquid biopsies, as it minimizes degradation and allows for more accurate detection in blood samples. In the realm of emerging biomarkers, such as Circulating tumor DNA (ctDNA) and microRNAs, hsa_circ_0052112 could complement these modalities by offering insights into tumor dynamics. While ctDNA found in the bloodstream and refers to DNA from cancerous cells and tumors provides information about the genetic alterations within the tumor 17, hsa_circ_0052112 may reflect the tumor's functional status and cellular processes more accurately. Furthermore, microRNAs are often less stable and can be influenced by various physiological states 18, whereas the stability of hsa_circ_0052112 could lead to more consistent and reliable measurements.
The in silco phase of our study indicated that the RNA-binding protein sites matching flanking regions of hsa_circ_0052112 are AGO2, EIF4A3, and PTB. Understanding the RNA-binding protein sites that correspond to the flanking regions of circRNAs is essential for comprehending their biogenesis, function, and role in disease, as well as for advancing research methodologies in molecular biology. Identifying RBP target sites is an important step toward understanding the mechanisms by which they conduct post-transcriptional regulation 19. AGO2 (Argonaute 2) is a vital protein taking part within the RNA-Induced Silencing Complex (RISC) and it plays a pivotal role in the regulation of gene expression through its function as an RNA-Binding Protein (RBP). It is well-known for its involvement in the mechanisms of miRNA and small interfering RNA (siRNA) pathways, facilitating the silencing of target mRNAs 20. AGO2 serves as a critical RBP for hsa_circ_0052112, influencing its function and stability and potentially playing a role in the regulatory networks involving miRNAs and gene expression.
EIF4A3 (Eukaryotic Translation Initiation Factor 4A3) is a member of the eIF4A family of RBPs and plays a critical role in the regulation of mRNA translation and stability. It was recently recognized as an oncogene. It is ordinarily recognized for its involvement within the Exon Junction Complex (EJC), that is vital for mRNA processing, transport, and translation 21.
Polypyrimidine Tract-Binding Protein (PTB), also known as PTBP1, is an RNA-binding protein that is involved in various processes, including splicing, mRNA stability, and translation. Analysis indicated that overexpression of PTBP1 generally predicted poor overall survival in patients with tumors such as liver hepatocellular carcinoma, adrenocortical carcinoma, lung adenocarcinoma, and skin cutaneous melanoma 22.
circRNAs are a group of endogenous RNAs that control gene expression at the transcriptional and post-transcriptional levels. Recent reports have indicated that circRNAs serve as innovative diagnostic biomarkers and favorable therapeutic targets for several cancer types by interacting with other non-coding RNAs such as miRNAs 23. The miRNAs are revealed as pivotal risk factors and regulatory elements in cancer by regulating the expression of their target genes. Genetic interactions between miRNAs and circular RNAs can generate complex regulatory networks with various carcinogenic processes that play significant roles in tumorigenesis, cancer development, and cancer progression 23. CircRNAs have recently been identified for their miRNA sponge function, which enables them to modulate the expression of miRNA target genes by acting as competitive endogenous RNAs (ce-circRNAs) 24.
In the in silco phase of our study, among the miRNAs that were introduced as targets of hsa_circ_0052112 by target scan, miR-194 was found to be associated with all subtypes of breast cancer even triple negatives 25,26. Studies have shown that miR-194 can act as a tumor suppressor in breast cancer by regulating the expression of target genes involved in cell cycle control and apoptosis 27.
On the other hand, miR-194 altered expression has been associated with poor prognosis in breast cancer patients. In some cases, downregulation of miR-194 has been linked to increased metastatic potential and resistance to therapy, implying that it may act as a potential biomarker for breast cancer progression and treatment response 28. miR-194 dual role as a potential tumor suppressor and its association with clinical outcomes highlights its importance in breast cancer research. In this research article, miR-194 is highlighted as a key microRNA involved in the post-transcriptional regulation of gene expression, playing critical roles in various biological processes such as cell proliferation, differentiation, apoptosis, and metabolism. Notably, as discussed earlier, miR-194 has been identified as a potential tumor suppressor in several cancers, including breast cancer. It can inhibit oncogene expression and promote apoptosis, thus influencing the balance between cell survival and death. Additionally, miR-194 regulates genes involved in cell adhesion, migration, and invasion, thereby affecting cancer metastasis 29.
In silco part of our study revealed that MAPK1, STAT1, TRAF6, and ERRB4 were identified as hub genes targeted by miR-194. Hub genes are central genes within a biological network that play a crucial role in regulating various cellular processes. They are often characterized by a high degree of connectivity, meaning that they interact with many other genes or proteins. In the context of gene networks, hub genes can influence the behavior of the network as a whole and are often implicated in critical biological functions, disease mechanisms, and responses to environmental changes. Identifying hub genes can be important for understanding complex biological systems and developing targeted medicine therapies.
The enrichment analysis identified significant associations between the hub genes and two critical biological processes: "PI5P, PP2A, and IER3 regulate PI3K/AKT signaling" and "negative regulation of the PI3K/AKT network". These findings underscore the potential roles of the hub genes in modulating the PI3K/AKT signaling pathway. For instance, MAPK1 is linked to cell proliferation and survival, suggesting its involvement in tumor growth and therapy resistance 30. STAT1, as a transcription factor, may influence the expression of genes that regulate the PI3K/AKT pathway, impacting cellular responses to disease signals 31. TRAF6 is known to activate AKT, promoting cell survival in cancer contexts 32, while ERRB4 is associated with signaling cascades that intersect with this pathway, particularly in breast cancer progression 33. The miR-194 interacts with the PI3K/AKT signaling pathway, a crucial cascade that regulates cell growth, survival, and metabolism. Activation of this pathway promotes cell survival and proliferation, often exploited by cancer cells, and is associated with chemotherapy resistance 34. The interplay between miR-194 and the PI3K/AKT pathway is significant in breast cancer, as low levels of miR-194 and aberrant activation of the PI3K/AKT pathway can serve as prognostic biomarkers linked to poorer outcomes and aggressive tumor characteristics.
Overall, the findings underscore the importance of miR-194 and the PI3K/AKT pathway in breast cancer biology, offering insights that could inform prognostic assessments and therapeutic strategies aimed at targeted interventions in breast cancer management. Additionally, hsa_circ_0052112 may modulate the PI3K/AKT pathway by acting as a sponge for microRNAs, thereby upregulating target hub genes, and influencing RNA-binding proteins and the transcription of nearby genes. The interactions between hsa_circ_0052112 and the hub genes could significantly affect breast cancer progression, as enhanced PI3K/AKT activation is commonly linked to increased cell proliferation, survival, and metastasis. These insights may inform future therapeutic strategies targeting the dysregulated PI3K/AKT pathway in breast cancer.
The phosphatidylinositol 3-kinase (PI3K)/Akt pathway plays a pivotal role in various cellular processes and is aberrantly engaged in cancers, contributing to the manifestation and progression of tumors. PI3K/AKT signaling pathway is a critical intracellular signaling pathway that regulates multiple cellular functions, including growth, proliferation, survival, and metabolism. This pathway is often activated in response to growth factors and hormones, playing a significant role in normal cellular processes 35.
The mechanism of action of PI3K enzymes PI3K/AKT signaling pathway is as follows: phosphorylate phosphatidylinositol to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3), which activates AKT, leading to its translocation to the plasma membrane and subsequent phosphorylation. This activation promotes cell survival, growth, and metabolism, thereby facilitating cell proliferation and inhibiting apoptosis, which can contribute to tumor growth, particularly in breast cancer 36. In hormone receptor-positive breast cancers, estrogen signaling can aberrantly activate this pathway, enhancing tumor growth and metastasis 37. As mentioned earlier The PI3K/AKT pathway is one of the most frequently over-activated intracellular pathways in several human cancers. This pathway, influencing various downstream target proteins, contributes to the carcinogenesis, proliferation, invasion, and metastasis of tumor cells. A multi-level impairment, including mutation, aberrant regulation of miRNAs, and aberrant phosphorylation of cascade factors, has been revealed in multiple cancer types. The deregulation of this pathway counteracts common therapeutic strategies and contributes to multidrug resistance. Interpretation of this pathway is essential for developing effective treatment strategies and personalized medicine approaches in breast cancer management 38.
The PI3K/AKT signaling pathway is crucial in breast cancer progression, affecting growth, survival, metabolism, and motility. It is activated by growth factors and hormones, leading to AKT activation, which promotes cell cycle progression and tumor growth while inhibiting apoptosis. This pathway supports the energy demands of rapidly proliferating tumor cells and enhances the tumor microenvironment through angiogenesis and metastasis. In hormone receptor-positive breast cancers, estrogen further stimulates this pathway, contributing to resistance against therapies like tamoxifen. Targeting the PI3K/AKT pathway is a focus of therapeutic interventions, although resistance is common, highlighting the need for combination therapies. Mutations in related genes, such as PIK3CA, are prevalent in breast cancer, underscoring the pathway's significance 39.
Conclusion :
The study highlights the significant upregulation of hsa_circ_0052112 in breast cancer patients, particularly in advanced stages and in association with specific receptor statuses, suggesting its potential role as a biomarker and therapeutic target. Additionally, the involvement of miR-194 and the PI3K/AKT signaling pathway underscores the complexity of gene regulation in breast cancer, emphasizing the need for further research to explore their implications in disease progression and treatment strategies.
While our study provides valuable insights into the role of hsa_circ_0052112, it is important to acknowledge certain limitations, including the relatively small sample size, which may affect the generalizability of our findings. Additionally, the absence of external validation in larger, more diverse cohorts restricts our ability to draw definitive conclusions regarding its clinical relevance. Future research should focus on expanding the sample size and conducting multicenter studies to validate our results in broader populations. Furthermore, exploring the therapeutic implications of hsa_circ_0052112 could lead to novel interventions, necessitating investigations into its functional mechanisms and potential applications in targeted therapies. This approach may enhance our understanding of its role in disease pathology and pave the way for innovative treatment strategies.
Acknowledgement :
We sincerely thank all the individuals who participated in this study. We would like to acknowledge Prof. Mozdarani and Prof. Kaviani for their support. This work was supported by a grant from NIGEB (Grant No. 805). The ethical code number is IR.MODARES.REC.1398.148.
Conflict of Interest :
No potential conflicts of interest were disclosed by the authors.
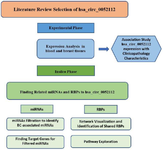
Figure 1. The flowchart of the research and step-by-step phases of the in silico phase of the study.
|
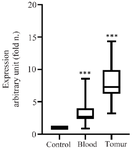
Figure 2. The mean expression of hsa_circ_0052112 in tissue and blood samples of breast cancer patients compared to normal control. Data were normalized by β-actin gene expression.
ANOVA test. p<0.0001.
|
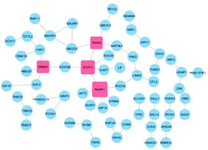
Figure 3. Gene Interaction Network and Hub Genes. The gene interaction network was constructed to identify hub genes. Nodes without any connections have been removed for clarity. Hub genes are indicated by squares, and specifically, genes with four or more interactions were selected as hub genes.
|
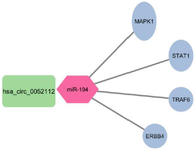
Figure 4. The interaction between has_circ_0052112_miR-194 and hub genes is visualized using Cytoscape.
|

Figure 5. Enrichment analysis of hub genes using the reactome database.
|

Table 1. Characteristics of breast cancer patients and controls
|

Table 2. Characteristics of circular RNA taken from the interactome site
|
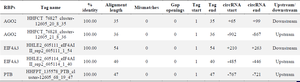
Table 3. RNA-binding protein sites matching flanking regions of circRNA hsa_circ_0052112
|

Table 4. List of miRNAs targeted by the TargetScan database for circular RNA hsa_circ_0052112
|

Table 5. Mean expression of hsa_circ_0052112 in different pathological conditions and hormone receptor status in breast cancer patients
p-value, and Effect size were calculated using T-test and Cohen's d test, respectively.
|
|