Monocytes and Macrophages as Unique Cellular Compartments Governing Non-Alcoholic Fatty Liver Disease and Inflammation
-
Hemmatian , Ghazale
-
Department of Immunology, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran
-
Rostamzadeh, Davoud
-
Medicinal Plants Research Center, Yasuj University of Medical Sciences, Yasuj, Iran
-
Baghaei, Kaveh
-
Gastroenterology and Liver Disease Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Iran
-
 Shabani, Mahdi
Department of Immunology, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran, Tel: +98 21 22439970; Fax: +98 21 22439970; E-mail: msshabani@sbmu.ac.ir
Shabani, Mahdi
Department of Immunology, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran, Tel: +98 21 22439970; Fax: +98 21 22439970; E-mail: msshabani@sbmu.ac.ir
Abstract: Non-Alcoholic Fatty Liver Disease (NAFLD) is a spectrum of liver diseases from simple steatosis to the most severe form of hepatocellular carcinoma. Liver injuries resulting from various factors, including viral infections, alcohol consumption, and metabolic disorders, trigger the activation of resident immune cells and the recruitment of circulating immune cells to the liver. This chronic inflammatory environment leads to tissue damage and the progression of liver fibrosis. Macrophages are highly versatile immune cells that play a dual role in fibrosis: they contribute to the progression of fibrosis (M1 and Ly6chigh macrophages) and its resolution (M2 and Ly6clow macrophages). M1 macrophages and those with high surface expression of Ly6C exhibit pro-inflammatory characteristics, while M2 macrophages and myeloid cells with low expression of Ly6C mitigate inflammation and inhibit fibrosis progression. Environmental stimuli influence the complex mechanisms hepatic macrophages regulate the fibrosis they encounter. Kupffer cells initiate the inflammatory cascade and recruit monocyte-derived macrophages, which modulate the propagation of fibrosis and promote fibrinolysis. Additionally, hepatic macrophages interact with other cell types through exosomes, facilitating the transfer of cellular components that influence the outcome of liver fibrosis. In this review, the critical role of macrophages in inflammation-induced fibrosis and tissue restoration is discussed.
Introduction :
Non-Alcoholic Fatty Liver Disease (NAFLD) is a long-lasting liver disease characterized by the accumulation of huge amounts of fat in the liver which can lead to liver-related death, especially among steatonecrosis and fibrosis-afflicted patients. NAFLD may progress to a more severe liver condition termed fibrosis 1. Liver fibrosis is the accumulation of extracellular matrix in response to repeated liver damage. It takes place when the capacity of the liver to substitute hepatocytes is overwhelmed and fibrillar collagen is abundantly accumulated 2. Cellular mechanisms contributing to the progression of liver fibrosis include the activation of Hepatic Stellate Cells (HSCs) and their trans-differentiation into myofibroblast-like cells, which produce large amounts of extracellular collagen and facilitate fibrosis progression 3.
Immune cells and liver fibrosis: Macrophages play contradictory roles in regulating liver fibrosis; they promote it through the production of IL-1β and TNF-α by M1 macrophages, while M2 macrophages can reverse liver fibrosis 4-7. Different subsets of T cells also influence the outcome of fibrosis: Th1 cells exhibit pro-inflammatory characteristics, Th2 cells release IL-13 and IL-4 to promote fibrogenesis, and Th17 cells produce IL-17, which activates HSCs and macrophages. In cases of chronic hepatic damage, despite the early apoptosis-inducing effects of Natural Killer (NK) cells on HSCs, these cells are ultimately considered to promote fibrosis. The changes in the number of immune cells in the context of liver fibrosis determine the persistence of inflammation. A decrease in NK cells and CD8+ cytotoxic T cells, along with an increase in CD4+ memory T cells, underscores the necessity of inflammation for the continuation of liver fibrosis 8. Hepatocytes secrete IL-8, CXCL1, and CXCL2 to promote the recruitment of neutrophils, which is followed by increased myeloperoxidase activity, reactive oxygen species production, and the formation of neutrophil extracellular traps, all contributing to the progression of inflammation. Dendritic cells, which perform antigen presentation to naïve T cells, play paradoxical roles in NASH, they release TNF-α, MCP-1, and IL-6 during the initial phases of liver damage to promote inflammation, but in the regression phases of liver injury, they facilitate fibrosis regression. Natural Killer T (NKT) cells, a component of acquired immunity, drive fibrosis progression through the secretion of IFN-γ, IL-4, IL-15, VCAM-1, CXCL16, and osteopontin, as well as through an overactive Hedgehog pathway. B cells exhibit dual roles in regulating liver inflammation by presenting oxidative stress-derived epitopes to CD4+ helper T cells and releasing IL-10 to inhibit CD8+ T cell-mediated inflammation. However, they also produce anti-oxidative stress epitopes, such as IgG, to mitigate inflammation 9. Dysregulation of matrix metalloproteinases, which promote matrix degradation, and tissue inhibitors of matrix metalloproteinases govern extracellular matrix remodeling, leading to the deposition of extracellular matrix components. Another contributing mechanism to liver inflammation and fibrosis is the disruption of immune tolerance mediated by regulatory T cells and dendritic cells, which instigate liver fibrosis 10. Despite the complex involvement of the immune system in liver inflammation, this study aims to briefly discuss the roles of macrophages. Kupffer cells, the resident macrophages of the liver, are responsible for maintaining liver homeostasis through the phagocytosis of apoptotic cells and pathogens. They mediate the liver's homeostatic state through immune regulation via IL-10 production, phagocytosis of gut-derived invading substrates and facilitating hepatic repair and regeneration. The production of TGF-β and PDGF by Kupffer cells leads to HSC activation. Furthermore, Kupffer cell-derived IL-6 and TNF-α contribute to the persistence of inflammation. Additionally, Kupffer cells are activated through the sensing of apoptotic hepatocyte signaling and NF-κB activation, promoting inflammation 4,5,7,11-13.
Liver macrophages, including Kupffer cells and monocyte-derived macrophages, constitute the hepatic macrophage pool. However, in response to chronic liver injury and inflammation, these macrophages adopt ambivalent functions, including those of M1 and Ly6chigh (lymphocyte antigen 6 complex, locus C1) monocytes. Following the cessation of the inflammatory response, macrophages acquire M2 and Ly6clow phenotypes to promote tissue repair 14. In mice, Ly6Chigh monocytes, which correspond to CD14high CD16low human monocytes, are considered pro-inflammatory. In contrast, Ly6Clow monocytes and their human counterparts, as CD14low CD16high monocytes, patrol along blood vessels and promote fibrosis regression due to their elevated expression of matrix metalloproteinases, phagocytosis-associated genes, and growth factors, extending beyond the M1 and M2 phenotypes 15. Traditionally, macrophage polarization states were categorized as M1 or M2. M1 macrophages (classically activated macrophages) are pro-inflammatory and contribute to tissue injury, while M2 macrophages (non-classically activated macrophages) are involved in tissue remodeling and efferocytosis, thereby inhibiting inflammation 16.
HSCs are non-parenchymal liver cells residing in the sub-endothelial space of Disse (also known as the perisinusoidal space) and interact with liver macrophages upon activation 17. Following we provide an update on the critical role of macrophages in liver fibrosis, as well as their interactions with other liver cells.
NAFLD spectrum and prevalence: NAFLD is a spectrum of liver disorders, ranging from Non-Alcoholic Fatty Liver (NAFL), and Non-Alcoholic Steatohepatitis (NASH) to liver fibrosis, cirrhosis, and Hepatocellular Carcinoma (HCC). Due to the strong association between NAFLD and type 2 diabetes, hyperlipidemia, obesity, and hypertension, NAFLD is considered a metabolic syndrome 18. NAFL is distinguished when the liver contains ≥5% steatosis without the presence of fibrosis. NAFL is usually benign and without significant risk of mortality. NAFL can progress to NASH (non-alcoholic steatohepatitis), characterized by at least 5% hepatic steatosis, accompanied by hepatocyte ballooning and liver inflammation. NASH may be followed by more advanced complications including liver fibrosis, cirrhosis, and HCC. When defining NAFLD, it must be considered that people who consume abundant quantities of alcohol must be excluded from this spectrum 19,20.
NAFLD imposes a great health burden on societies worldwide. Based on a research study conducted by Ge et al, it was estimated that NAFLD prevalence would increase dramatically from 1990 to 2017 with the highest prevalence observed in many territories including East Asia and the Middle East 21. Furthermore, NAFLD prevalence and mortality highly increased in the United States 22,23. These findings underscore the critical need for healthcare professionals to implement thorough measurements in managing this disease.
Inflammation initiation: Diverse innate and adaptive immune cells are implicated in liver fibrosis. The innate immune cells of the liver serve as the first line of defense against invading injuries. However, when chronic damage takes place, Endoplasmic Reticulum (ER) stress (unfolded protein response), Reactive Oxygen Species (ROS), and lipid overload especially free fatty acids, induce liver inflammation initiation. A disruption occurring in the equilibrium of ROS production and antioxidant mechanisms causes molecular damage and leads to fibrosis development 24-27. The main instigators of liver inflammation are attributed to several factors including Free Fatty Acids (FFAs) and bacterial-derived lipopolysaccharide that activate immune cells through interaction with Damage-Associated Molecular Patterns (DAMPs) and Pathogen-Associated Molecular Patterns (PAMPs), respectively leading to initiation of liver inflammation. Gut dysbiosis and adipose tissue dysfunction accelerate inflammation progression 28,29. Furthermore, leukocytes recruitment (macrophages, neutrophils, T cells) trigger tissue damage in NAFLD 30-32. Hepatocyte cell death is mediated by oxidative stress and FFAs deposition which are a result of mitochondrial dysfunction observed in NAFLD 28,30. In addition, an inequity between reactive nitrogen species and reactive oxygen species leads to fibrosis 33.
The liver resident macrophages termed Kupffer cells are the most abundant resident macrophages in this organ. They are hepatic sentinels against exogenous and endogenous danger signals. They are characterized as F4/80+, CD11bintermediate, CD45+, and C-type lectin domain family 4, member f (CLEC4F) positive cells. They become activated after recognizing Lipopolysaccharide (LPS) by toll-like receptor 4 (TLR4) and transmitting signals through myeloid differentiation primary response protein 88 (MyD88) dependent or MyD88 independent pathways, thereby initiating the inflammatory cascade and activation of HSCs 14,24. Macrophages acquire the pro-inflammatory phenotype by ROS production, phagocytosis, elevated expression of adhesion molecules, and cytokines to decrease danger signals. Macrophages' anti-inflammatory phenotype secrete TGF-ꞵ, and IL-10 and accomplish apoptotic cell phagocytosis to decrease inflammation. However, as a result of persistent inflammation, inflammatory monocytes secrete TGF-ꞵ to mediate the activation of fibroblasts. M2 macrophages are fibrogenic subsets driving fibrosis perpetuation via profibrotic mediators and matrix metalloproteinases prevention (Figure 1) 34-36.
In addition, other metabolites such as adipokines, and cholesterol through scavenger receptors A and CD36, and DAMPs e.g., ATP, mitochondrial DNA, and high mobility group box 1 originated from injured hepatocytes and free fatty acids via TLRs induce the activation of Kupffer cells 37,38. Activated Kupffer cells produce transforming growth factor-ꞵ1 (TGF-ꞵ1), platelet-derived growth factor (PDGF), interleukin-1 (IL-1), monocyte chemoattractant protein 1 (MCP1), IL-12 and tumor necrosis factor-alpha (TNF-α) essential for HSCs proliferation and recruitment. Kupffer cells gelatinases and chitotriosidase expression also induce collagen type I production and HSCs activation, respectively. Kupffer cells-derived TGF-ꞵ1 also stimulates the differentiation and activation of fibroblasts 39. In addition, activated Kupffer cells also serve as a source of IL-6 and inducible Nitric Oxide Synthase (iNOS) expression. Indeed, iNOS stimulates Nitric Oxide (NO) production which together with ROS produce Reactive Nitrogen Species (RNS) and stimulates the HSCs activation 40. The reciprocal interplay between hepatic macrophages and HSCs perpetuates fibrosis. Indeed, Kupffer cells assist HSCs activation, survival, and pro-fibrogenic phenotype maintenance through TGF-ꞵ, oncostatin M, PDGF, TNF-a, IL-1ꞵ, TNF superfamily member 12 (TNFSF12A) and Epidermal Growth Factor Receptor (EGFR) ligand secretion. In turn, activated HSCs release C-C motif chemokine ligand 2 (CCL2), hyaluronan, IL-6, and TGF-ꞵ which are essential for monocytes recruitment, activation, and polarization. Activated HSCs also contribute to inflammation persistence through secreting CCL21 and IL-1ꞵ. CCL21 helps to recruit immune cells to sites of inflammation. In addition, NLRP3 inflammasome activation leads to IL-1ꞵ secretion that assists inflammation perpetuation 41,42. This positive feedback loop preserves hepatic inflammation and fibrosis 43. Furthermore, Kupffer cells-derived CCL2 and CCL5 contribute to Ly6chigh monocytes infiltration to the liver that promotes hepatic inflammation (Figure 1) 13.
Inflammatory monocytes/macrophages in fibrosis progression: Kupffer cells TLR signaling, and activation cause upregulated levels of CCL2 or MCP1 production. These mediators induce the recruitment of Monocyte-derived Macrophages (MoMFs) which are characterized by Ly6c, C-X3-C motif chemokine receptor 1 (CX3CR1), C-C chemokine receptor 2 (CCR2), and CD11b expression and accomplish macrophage replenishment upon injury. Based on their Ly6c expression, Ly6chigh monocytes equal to CD14high CD16low human monocytes contribute to inflammation propagation 44,45. A research study conducted by Baeck et al deduced CCL2 as a chemokine indispensable for Ly6chigh pro-fibrogenic monocytes infiltration that hinders fibrosis resolution 46. This subset is crucial for determining fibrosis fate; because they are characterized as the main proliferative macrophage subset expressing iNOS 47.
Macrophages adopt different phenotypes depending on their microenvironmental stimuli. Interferon-Ƴ (IFN-Ƴ), LPS, and IL-12 induce M1 macrophages polarization which is characterized by the production of iNOS, TNF-a, IL-1ꞵ, IL-6, IL-12, IL-8, and IL-15 and participate in pathogen elimination and inflammation propagation. These macrophages, also known as classically activated macrophages, are distinguished by their cell surface expression markers, including CD86, CD80, TLR4, TLR2, Interleukin-1 Receptor (IL-1R), and major histocompatibility complex class II (MHC-II). In addition, they secrete chemokines like CCL5, CCL2, CCL3, CCL4, C-X-C motif ligand 8 (CXCL8), CXCL9, CXCL10 and CXCL11. M1 macrophages also trigger HSC activation by secreting TGF-ꞵ and facilitating fibrosis progression 48-51.
Restorative monocytes/macrophages in fibrosis regression: Macrophages are highly plastic immune cells capable of both promoting and reversing liver inflammation and fibrosis. Ly6chigh monocytes can polarize to Ly6clow monocytes equal to CD14low CD16high human monocytes that are implicated in fibrosis regression 44. These monocytes have been shown to contribute to fibrosis regression by matrix metalloproteinases MMP9 and MMP12 expression and induction of phagocytosis of cellular debris namely efferocytosis. Ly6clow monocytes exhibit anti-fibrotic characteristics corroborated by the observation that they express CD47/Macrophage Migration Inhibitory Factor (MIF) and Insulin-like Growth Factor 1 (IGF1) 15.
M2 macrophages also known as non-classically activated macrophages are stimulated by IL-13, IL-10, and IL-4. M2 macrophages express CD163, CD206, dectin-1, scavenger receptors and CCR2. Matrix metalloproteases expression by M2 macrophages facilitates wound healing. TGF-ꞵ, arginase-1 (Arg1), and IL-10 produced by M2 macrophages contribute to anti-inflammatory characteristics 48. In addition, in a research study conducted by Wan and colleagues, it was reported that M2-polarized Kupffer cells induce M1 Kupffer cells apoptosis to inhibit inflammation 52. In general, M2 macrophages are anti-inflammatory due to IL-13, IL-4, IL-10, and TGF-ꞵ secretion, and assist wound healing 50. Due to the high versatility of M2 macrophages, they are further subdivided into M2a, M2b, M2c, and M2d. M2a and M2b macrophages regulate immune responses, and M2c macrophages promote wound healing and tissue remodeling. In addition, this subgroup of macrophages suppresses immune responses. M2d macrophages are implicated in angiogenesis and assist tumor development 49. Due to the aforementioned dual paradoxical characteristics of macrophages in exacerbating and ameliorating liver inflammation and fibrosis, an approach that targets inflammatory macrophages or promotes the polarization of restorative macrophages holds great potential in mitigating the disease.
Macrophages therapeutic targeting: There have been several strategies proposed to mitigate liver fibrosis. The primary therapeutic approaches focus on reducing the activation of Kupffer cells through modulation of the gut-liver axis. These strategies include preventing monocyte recruitment via CCL2-CCR2 inhibition, directing macrophage polarization from a pro-inflammatory to a restorative phenotype, and the transplantation of autologous monocyte-derived macrophages as well as bone marrow-derived macrophages. All of these strategies aim to alleviate liver inflammation and fibrosis 6,13,45,53,54.
Macrophages are compelling targets for liver inflammation, as they play crucial roles in the progression and regression of liver fibrosis. Therefore, any agent that targets macrophages may have the potential to alleviate liver fibrosis. A summary of some pharmacological interventions is presented in table 1.
Exosomes-mediated cellular crosstalk in liver fibrosis: The interaction between monocytes/macrophages and other cell types plays a crucial role in determining the fate of fibrosis. Liver sinusoidal endothelial cells express delta-like canonical Notch ligand 4 (DLL4), and the signaling pathways involving Notch, TGF-β, and LXR promote the trans differentiation of monocytes into Kupffer cells, which are recruited to the injured liver to expedite fibrosis progression. Furthermore, HSC-derived IL-1β and TNF-α enhance the differentiation of monocytes into Kupffer cells. Macrophages release PDGF, TGF-β, and ROS, which induce HSC activation. In turn, HSCs release CCL2 to promote the infiltration and activation of macrophages. This creates a positive feedback loop in which macrophages support HSC survival, while HSCs inhibit macrophage phagocytosis, thereby perpetuating inflammation 43,65. Additionally, macrophages facilitate Endothelial-to-Mesenchymal Transition (EndMT) in endothelial cells, a process characterized by increased invasiveness and migration, cytoskeletal reorganization, and loss of cellular adhesion, all of which contribute to HSC activation. Endothelial cells upregulate Vascular Adhesion Protein-1 (VAP1) and the integrin α4 subunit VLA-4, further stimulating monocyte recruitment. Macrophages, in turn, mediate endothelial disruption 43,65. Despite the complex interplay between macrophages and various immune cells that govern inflammation, the following section aims to elucidate macrophage-mediated communication through exosomes. Reason: Improved clarity, vocabulary, and technical accuracy while correcting grammatical and punctuation errors.
Due to the importance of secreted exosomes in cell-cell communications, in the following section, the effect of exosomes derived from either macrophages or other cellular sources on liver fibrosis outcome will be explained. Exosomes are types of extracellular vesicles ranging in size from 30-100 nm which contain multiple types of cargo including proteins, lipids, and nucleic acids, and assist intercellular communication. Exosome cargoes (e.g., miRNAs) influence the recipient cells and may stimulate or inhibit liver fibrosis 66-68. Chen et al showed that exosomes derived from THP-1 (human leukemia monocytic cell line) macrophages treated with LPS altered miR-103-3p and could therefore stimulate HSCs activation and proliferation 69. Jiayi et al observed that LPS-mediated stimulation of THP-1 cells promotes exosomes secretion rich in miR-155-5p which elevates oxidative stress, collagen synthesis as well as migration and proliferation of HSCs 70. These studies corroborate macrophage-derived exosomes in the progression of hepatic fibrosis. In another study by Wan et al, they showed that miR-411-5p in M2 macrophages-derived exosomes impeded HSC activation through Calmodulin-regulated spectrin-associated protein 1 (CAMSAP1) inhibition and the ensuing down-regulation of collagen and α-smooth muscle actin (α-SMA) in HSCs 71. Liu and coworkers reported that miR-192-5p expression in exosomes originated from lipotoxic hepatocytes stimulated pro-inflammatory M1 macrophages polarization via Rictor (rapamycin-insensitive companion of mammalian target of rapamycin)/Akt/FoxO1 (forkhead box transcription factor O1) pathway 72. It has been shown that lipids-mediated death receptor 5 induction in hepatocytes assists extracellular vesicles secretion which in turn instigates pro-inflammatory macrophage polarization 73. Lysosomal dysfunction caused by cholesterol mediates hepatocytes exosomal miR-122-5p release that induces M1 macrophages polarization and increases inflammation 74.
According to Chen and co-workers research study, LPS-induced macrophage activation and exosomal miR-500 release led to HSCs activation and proliferation in CCl4 (carbon tetrachloride)-induced mice liver fibrosis model through MFN2 targeting 75. Benbow and colleagues also confirmed that exosomes derived from activated HSCs stimulated macrophages' inflammatory TNF-α and IL-6 expression and migration 76. Moreover, miR-148a in mesenchymal stem cells-derived exosomes facilitates macrophage phenotype switching from the pro-inflammatory M1 to anti-inflammatory M2 subset by Kruppel-Like Factor 6 (KLF6) and signal transducer and activator of transcription 3 (STAT3) inhibition transcription factors 77. In conclusion, the source of exosome-producing cells in different microenvironments causes the alteration of miRNAs contents which induces fibrosis induction or restoration.
Conclusion :
Macrophages are regarded as the most critical immune cells in the regulation of liver fibrosis. Different subsets of pro-inflammatory and anti-inflammatory macrophages play opposing roles in the progression and resolution of liver fibrosis. Therefore, strategies that inhibit the harmful effects of inflammatory monocytes, while promoting restorative monocytes or targeting their intercellular communication, could be promising avenues for alleviating the disease. However, further in-depth studies are necessary to identify the various monocyte subsets and the cellular pathways involved in fibrosis.
Acknowledgement :
This work was financially supported by Shahid Beheshti University of Medical Sciences (grant number: 43008191).
Conflict of Interest :
Authors declare no conflict of interest.
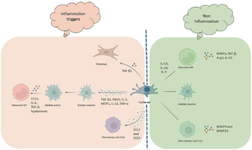
Figure 1. Monocyte and macrophage of the liver in the progression and restoration of fibrosis.
Kupffer cells become activated after recognizing LPS, adipokines, cholesterol, DAMPs, and free fatty acids. Activated Kupffer cells produce TGF-ꞵ1, PDGF, IL-1, MCP1, IL-12, and TNF-α essential for HSCs proliferation and recruitment. Kupffer cells-derived TGF-ꞵ1 also stimulates the differentiation and activation of fibroblasts. The reciprocal interplay between hepatic macrophages and HSCs perpetuates fibrosis. Indeed, Kupffer cells assist HSCs activation, survival, and pro-fibrogenic phenotype maintenance through TGF-ꞵ, oncostatin M, PDGF, TNF-a, IL-1ꞵ, TNFSF12A, and EGFR ligand secretion. In turn, activated HSCs release CCL2, hyaluronan, IL-6, and TGF-ꞵ which are essential for monocytes recruitment, activation, and polarization. Furthermore, Kupffer cells-derived CCL2 and CCL5 contribute to Ly6chigh monocytes infiltration to the liver that promotes hepatic inflammation. In normal conditions, Ly6clow monocytes have been shown to contribute to fibrosis regression by matrix metalloproteinases MMP9 and MMP12 expression and induction of phagocytosis of cellular debris namely efferocytosis. M2 macrophages are stimulated by IL-13, IL-10, and IL-4. Matrix metalloproteases expression by M2 macrophages facilitates wound healing. TGF-ꞵ, arginase-1 (Arg1), and IL-10 produced by M2 macrophages contribute to anti-inflammatory characteristics.
|
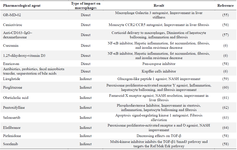
Table 1. Therapeutic agents targeting macrophages
|
|