Opportunistic Challenges of Computer-aided Drug Discovery of Lipopeptides: New Insights for Large Molecule Therapeutics
-
Yadav, Manisha
-
Department of Biotechnology, National Institute of Technology Raipur, C.G, India
-
 Eswari, J. Satya
Department of Biotechnology, National Institute of Technology Raipur, C.G., India, Tel: +97 52135824; E-mail: satyaeswarij.bt@nitrr.ac.in, eswari_iit@yahoo.co.in
Eswari, J. Satya
Department of Biotechnology, National Institute of Technology Raipur, C.G., India, Tel: +97 52135824; E-mail: satyaeswarij.bt@nitrr.ac.in, eswari_iit@yahoo.co.in
Abstract: Computer-aided drug designing is a promising approach to defeating the dry pipeline of drug discovery. It aims at reduced experimental efforts with cost-effectiveness. Naturally occurring large molecules with molecular weight higher than 500 Dalton such as cationic peptides, cyclic peptides, glycopeptides and lipopeptides are a few examples of large molecules which have successful applications as the broad spectrum antibacterial, anticancer, antiviral, antifungal and antithrombotic drugs. Utilization of microbial metabolites as potential drug candidates incur cost effectiveness through large scale production of such molecules rather than a synthetic approach. Computational studies on such compounds generate tremendous possibilities to develop novel leads with challenges to handle these complex molecules with available computational tools. The opportunities begin with the desired structural modifications in the parent drug molecule. Virtual modifications followed by molecular interaction studies at the target site through molecular modeling simulations and identification of structure-activity relationship models to develop more prominent and potential drug molecules. Lead optimization studies to develop novel compounds with increased specificity and reduced off targeting is a big challenge computationally for large molecules. Prediction of optimized pharmacokinetic properties facilitates development of a compound with lower toxicity as compared to the natural compounds. Generating the library of compounds and studies for target specificity and ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) for large molecules are laborious and incur huge cost and chemical wastage through in-vitro methods. Hence, computational methods need to be explored to develop novel compounds from natural large molecules with higher specificity. This review article is focusing on possible challenges and opportunities in the pathway of computer-aided drug discovery of large molecule therapeutics.
Introduction :
Exceptionally diversified bioactivity of natural pro-ducts leads to the high demand to explore them as potential drugs. Limited accessibility of natural products is often replaced with organic synthesis. As it is well recognized, natural compounds of complex structures are difficult to derive through pure chemical synthesis. Semi-synthesis approach solves to retain the original activity of natural products with certain chemical modifications which also enables the bioactive derivatives into more drug-like compounds 1,2. Hence, novel derivatives are developed in order to yield potential compounds with better medicinal value.
Predominantly, natural compounds with structural modifications are performed in an attempt to achieve the target selectivity, better potency and additional properties of the drug. Small molecule therapeutics are generally found with the molecular eight less than 500 Dalton and follows the Lipinski’s rule of 5. While large molecules are found with molecular weight above 500 Dalton. Various microbial peptide-based large molecules such as lipopeptides and glycopeptides are natural bioactives which are produced as microbial metabolites and are found with various medicinal properties such as antifungal, antibacterial, antimicrobial, antitumor, antiviral, antiparasitic and hemolytic activities 3,4. One such example of an emerging peptide therapeutics is lipopeptide-based drugs 5. These are microbial by-products of natural origin.
Lipopeptides are produced as secondary metabolites majorly from the bacteria of Bacillus species 6,7. These are amphiphilic compounds with one hydrophobic fatty acid tail and a cyclic hepta to deca peptide core with a molecular weight of 1-1.5 kDa 8-11. Structural modification in microbial lipopeptides can enable these natural compounds for desired and selective drug-like derivatives. Computer-aided drug designing of such large molecules have not been explored much because of their complex structure 9,12. Computational drug discovery is a cost-effective way to understand the drug designing at the molecular level. Several studies have been reported on the drug discovery of small molecules and various capable in silico methods are also available in order to develop the optimized drug molecules with improved specificity for the target proteins of a particular disease 13. These results are validated and closely related to what has been developed in vitro but in silico methods narrow down the research process through virtual screening because these can give insight into the structure-activity relationship of the drug molecule 14,15. Such in silico methods have certain limitations when it comes to large molecule-based drugs such as peptides, peptidomimetics and proteinogenic pharmaceuticals 16. Although such big structures of peptide-based drugs bring more opportunistic probability to designing more specific drug molecules because these large molecules are available with various functional groups to lie upon and tweak the structure so as to fit into the binding pocket of the target protein 17,18.
This leads to increased specificity and lowers the off-targeting of the drug molecules which is a common problem with small molecule-based drugs and subsequent side effects of off-targeting can also be taken care of 19. Utilizing large molecules in drug discovery bring lots of challenges and opportunities have been explored in this review article. Development of in silico methods and various computational tools are of great help in bringing such complex molecules in the drug discovery pipeline. Such techniques as molecular docking gives deeper insight at the molecular level so that to develop more potent drugs for the specific targets.
Complex structures of peptide-based drugs give more opportunity to play with the structure for the drug discovery researchers look for the critical component of drug activity which are called the pharmacophore. Retaining the pharmacophore of the drug and addition of potential functional groups for improved drug efficacy are now possible with computer-aided drug design. Here the utilization of natural compounds such as microbial peptides such as lipopeptides and glycopeptides serve as the template for designing the novel drug derivatives through a various structure-based and analogue-based drug designing 20. Analogue-based drug discovery is a semisynthetic approach where in novel analogs of natural compounds are targeted as prospective drug candidates. It is cost-effective, environment friendly, safe and less toxic approach than using the synthetic approach of designing and production of synthetic peptide-based drugs which are expensive and require tedious lab work in designing such complex compounds 21.
Requirement of in-depth studies on computer-aided drug discovery of large molecules is indicative because no such studies have been reported specifically dedicated to challenging opportunities while working on large molecule therapeutics. Peptide therapeutics have been explored with in-vitro studies well in combating drug resistance in cancer treatment. But there is a need to explore large molecule compounds through natural origin through computational methods. Hence, we have put together the consolidated studies on large molecule therapeutics while working with in silico methods. Target specificity is challenging with small molecules which is attainable with large molecule therapeutics. This review article will be of help to the researchers working on drug resistance in cancer treatment and infectious diseases to accelerate their studies in the direction of utilizing computational techniques for developing potential drugs in a cost-effective and environment friendly manner.
Lead optimization of a large molecule is a laborious task that includes multiple chains of chemical reaction with a low probability of success. Conclusively it is a hit and trial method, which is a reason for failure in defeating drug resistance. Hence, the coupling of computational tools for the identification of structure-activity relationship studies, attaining target specificity and analyzing the receptor-ligand interaction can be done in a synchronized way to generate more prominent novel lead to test clinically 23.
Peptide therapeutics: Development of peptide therapeutics has achieved wide interest because of their being prospective drug candidates. These are regarded to be safe, highly selective, and efficacious with better tolerability and exhibit a promising pharmacological profile 29,30. Production of
peptide-based pharmaceuticals are consequently more economical because of the lesser production complexity owing to their inherently smaller size as compared to the protein therapeutics. Hence, they are found more agility in their pharmacokinetic properties at the same time. The abovementioned features distinguishing properties which place them apart from protein-based biopharmaceuticals and small molecule drugs 31. So far, there have been existence of around 7000 peptides of natural origin which have been shown to exhibit a broad range of bioactivities such as tumour homing, antiparasitic, antiviral, antihypertensive, antiangiogenic, antimicrobial, antibiofilm, anticancer, etc. Consequently, these can be targeted against various diseases like diabetes, cancer, cardiovascular diseases, etc 32-34. Till now, approximately 60 peptide-based drugs have been approved by FDA 35, whereas 150 more peptides are currently in the preclinical and clinical studies. Research interest in peptide therapeutics have been increased due to high specificity and relatively lower toxicity derived from tight binding to the targets. However, understanding of the molecular mechanism of protein-peptide interaction has important implications on understanding the pharmacology of drug 36.
Nowadays, peptide-derived drugs are receiving much attention as promising drug candidates due to the opportunity in the rational design of novel derivatives. Rational design of derivatives of peptide therapeutics generally require characterization of the underlying interaction of protein and a peptide drug at the structural level 37. The challenge with such large molecule therapeutics is difficult in conducting structural characterization, hence reliable and powerful computational tools are required. In recent years, various approaches have been developed for the understanding of protein-peptide interaction at the molecular level by peptide docking. This helps in the prediction of protein-peptide complex structure and this also includes varying degrees of information about its binding conformations. Computational methods such as fragmented docking and incremented docking can resolve the visualization of interaction of peptide-based drugs at the molecular levels 38.
Lipopeptides: Lipopeptides are microbial metabolites that are majorly produced by Bacillus species. These are amphiphilic compounds composed of one cyclic peptide moiety of seven to ten residues and a fatty acid side chain. The molecular weight of microbial lipopeptides is approximately 1-1.2 kDa. These are known for their broad-spectrum activity as pore-forming, antimicrobial, antiparasitic, anti-tumor, biosurfactant, anti-obesity and antiviral properties. Daptomycin is the first FDA-ap-proved lipopeptide known. Similarly, surfactin, fengy-cin, iturin, tsushimycin, polymyxin, vancomycin and aspartocin are a few examples of microbial lipopeptides with antimicrobial and anticancer properties 8,22,23.
Discussing physico-chemical properties and structure of a large molecule surfactin, which is an example of amphiphilic cyclic lipopeptide, it constitutes a heptapeptide (ELLVDLL) wherein the chiral sequence LLDLLDL is interlinked with a 12-16 carbon long β-hydroxy fatty acid chain to form a lactone ring structure (Figure 1). Position of hydrophobic amino acid residues are 2, 3, 4, 6 and 7. While two negative charges are introduced through glutamyl and aspartyl residues at position 1 and 5, respectively. Generally, various surfactin isoforms are also found as a mixture of peptide derivatives with a varied length of aliphatic chain in the cell 11,12,24. Surfactin adopts a β-sheet structure with a horse-saddle structure characteristically, probably which is responsible for its biological activeties of the broad spectrum. Table 1 is depicting a list of peptide therapeutics with their applications and mechanism of action 6,23,25-28.
Short cationic peptides: Antimicrobial cationic peptides are produced by various organisms, plants and insects. These are majorly produced as a part of the immediate effect of a non-specific defense mechanism against infections. There is an emergence of developing novel class of antibiotics, due to the increasing antibiotic resistance amongst bacterial pathogens. Hence, cationic peptides are discovered and developed as new class of anti-infective agents through clinical trials 39. Apart from the antimicrobial properties of such peptides are capable to play an effective role in innate immunity and to regulate the gene expression in eukaryotic cells, hence can be explored for diseases like cancer 40,41.
Cyclic peptides: Polypeptide chain with a circular bond sequence is called cyclic peptides. This is through the bonds between the carboxyl and amino group of peptides, for example cyclosporin, bacitracin, colistin, amanitin, etc. Many cyclic peptides have been developed in laboratories and many are found in nature. The length of such peptides varied from 2 to 100 residues. They are frequently antimicrobial and toxic in nature. These properties have various medical applications as antibiotics, immunosuppressive agents 42. Generally, the thin layer chromatography technique is used for the detection of cyclic peptides in crude extracts of biomass 43.
Advantages of microbial peptides: Cyclization of peptide for structural rigidity: Beyond Lipinski’s rule of five. The biggest challenge of using peptide-based drugs including cyclic peptides and short cationic peptides, these large molecules distinctively disobey the Lipinski’s rule of five. According to which such drugs are generally of higher molecular weight mostly more than 500 Dalton. Another challenge needs to be considered is, the presence of free rotatable bonds which as per the rule of 5 limits to the presence of 10 rotatable bonds. However, the peptide-based drugs are abandoned with a greater number of free rotatable bonds. This challenge can be managed by the cyclization of the structure which is feasible with cyclic peptides 44. The reason the presence of a greater number of free rotatable bonds in drug molecules can make complex with surrounding multiple water molecules. This leads to the poor absorption of drugs in the gut and difficulty reaching the systemic circulation. Hence, cyclization of structure makes the less availability of free rotatable bonds and brings more rigidity constraint in the structure of the drug 45,46. More number of the hydrogen bond donor and acceptor group in the peptide-based drugs give many opportunities to develop the drug molecule with increased specificity. The drug molecule can be designed to obtain the desired bonds to occupy the deeper cavities of the binding pocket to inhibit the specific target protein 182 17,47.
Combating multidrug resistance with large molecules: Peptide-based drugs have revolutionized the fight against drug resistance be it the category of antimicrobial drugs and anticancer drugs. Such large molecule drugs are of great interest because there is a huge opportunity to tweak the structure to design the drug molecule with more favorable features to combat multidrug resistance 48. The surface-active properties of amphiphilic lipopeptides have already proven to act upon the cell wall of microbes and the mechanism of action to disrupt the cell wall, hence are more potent against the drug resistance 49,50.
There is a definite need to work more efficiently on the structure-activity relationship properties of lipopeptides so that to understand what mechanism is responsible for the toxicity and how the efficacy of such drugs can be improvised 51. Large molecules like lipopeptides exhibit the mechanism of action as the effective surface-active compounds, because of being an amphiphilic molecule. Hence their pore-forming capacity can disrupt the cell membrane and this leads to cell death 52,53. This mechanism is a strength of such compounds which are not only target driven. However, a mutation in the target protein is the biggest reason for drug resistance in infectious diseases as well as in cancer treatment. Alternative therapeutics with surface active properties can be advantageous in combating multi-drug resistance and keep them into the category of last resort antibiotics 54. Apart from this docking studies have proven the ability of large molecular inhibitors to dock the target protein at the multiple sites (Figure 2). The opportunity with improved selectivity for membranes can be attained with detailed structure activity relationship studies with the help of powerful computational methods.
Lipopeptides as potential anticancer agents: Microbial lipopeptides produced from Bacillus subtilis are found with anticancer properties which includes inhibitory activity in vitro and in vivo against human breast cancer via the disruption of AKT-ERK pathway. The induction of apoptosis for melanoma skin cancer A375 cells have been reported which suggests the activity through an interaction of lipopeptide molecules with the plasma membrane and the K562 cells lines of leukemia associated with PARP (poly ADP-ribose protein), caspase-3 is also found sensitive against lipopeptides based anticancer studies. The lipo-peptide compounds are composed of a hydrophobic fatty acid chain and hydrophilic cyclic peptide moiety which make it a compound of interest from natural origin majorly from Bacillus species bacteria. These are found in various pharmaceutical applications as antifungal, antiviral, anticancer, antimicrobial, anti-obesity, immunomodulators, plant inducers, etc. Many of them have been explored as antiviral agents and against SARS-Cov2 as well.
Iturin, surfactin and fengycin are the major Bacillus lipopeptides have been reported to be found with anticancer activities on extensive research. The suppression of the proliferation of human colon carcinoma on LoVo cell lines, suppression of TPA-induced invasion of breast cancer through inhibiting the expression of MMP9 and killing of MCF7 cell lines for human breast carcinoma through ROS/JNK mediated caspase/mitochondrial pathway is reported. Fengycin is reported to be active against 95D cell lines of non-small lung cancer 55.
Owing to the potential effect of lipopeptides on cancer, various studies have been reported to decipher the mechanism of action of such large molecule therapeutic options. Exploring such compounds through computational studies will further aid-in for depiction of interaction, visualization of the complex with a receptor, understating the energies and exploring the opportunities for lead optimization and novel drug discovery from the compounds of natural origin 56.
Opportunistic challenges with computational drug discovery: Drug design is a process which involves the finding of new drugs through an inventive process based on the structural knowledge of the biological target 57. Mostly, drugs are small organic molecules that take part in the inhibition and activation of the function of biomolecular target i.e., protein, resulting in a therapeutic benefit to curing a disease. Basically, drug design deals with predicting and designing molecules with complementary shape and charge for the target biomolecule to act upon and bind for therapeutic benefit. Drug design with aid of computer modeling techniques is referred to as computer-aided drug design 58,59. Drug design that incurs the known three-dimensional structure of the target protein is called as structure-based drug design 59. Biopharmaceuticals including peptide therapeutics are generally large molecular compounds explored as drugs that are generally derived from microbial sources and synthetic approaches 60. Chemical synthetic approach involves multiple steps of chemical reactions to obtain the final product which is an expensive, tedious and not an environment-friendly approach. On the other hand, computational drug discovery and designing of large molecule-based drugs is a challenging approach with a lot of opportunities in structural modification to obtain specificity for target and the disease. These kinds of tasks incurred huge money when it comes to in vitro and in vivo studies 13.
Computational studies can be of great help for initial screening, structural modification and identifying structure-activity relationship at the molecular level. Hence, the approach remarkably reduces the cost of drug discovery, if efficient in silico techniques and methods are employed. The fundamental goal of drug design is the prediction of binding interaction of given molecule and target and how strong the interaction is. Most often, molecular dynamics and molecular mechanics is used for the estimation of the strength of intermolecular interaction. These methods are also used for the prediction of conformations and changes in the conformation of the method when the interaction takes place 60. Ab initio, semi-empirical, quantum chemistry and density functional theory are the most sought methods used to generate optimized parameters for the calculations of molecular mechanics. These methods also provide an estimation of electronic properties such as polarizability and electrostatic potential etc. that influence the binding affinity of the candidate drug 61.
Computer-aided drug design is used in the following protocol of drug discovery (Figure 3):
1. Hit identification-Virtual screening (structure-based/ ligand-based drug design)
2. Hit to lead-Optimization of selectivity and affinity (structure-based drug design, QSAR, etc.)
3. Lead Optimization of other pharmaceutical properties while maintaining affinity 13.
Structure-activity relationship (SAR/QSAR): Relationship of the chemical structure of a compound or molecule with its corresponding activity is studied as the Structure-Activity Relationship (SAR). SAR analysis determines the chemical group which is responsible for a specific biological effect of the drug. This enables the effective structural modification for the improved potential of a bioactive compound (a drug) by altering the chemical structure. Medicinal chemist often uses chemical synthesis technique for the insertion of new chemical groups into the biochemical compound and further testing of modifications for the biological effects are done 62,63. Large molecules such as cyclic lipopeptides come with huge opportunity for structural modification to improve the specificity towards the target protein. The opportunity with the help of SAR studies can be explored using various software available for 2D, 3D and field-based QSAR studies to explore the desired properties of drugs 64. Studies based upon chromatographic indices and High Performance Liquid Chromatography (HPLC’s) are used to predict the antibacterial activity and lipophilicity of short cationic lipopeptides respectively. The results obtained using Quantitative Structure Toxicity Relationship (QSTR) and Quantitative Structure Activity Relationship (QSAR) analysis, the models indicate the importance of lipophilicity. Since, lipophilicity is one of the vital physiochemical parameters determining the biological properties for xenobiotics. The crucial conclusion elucidated through such studies is that the lipopeptides exhibit nonspecific interaction between bacterial membranes and erythrocytes. On the basis of various affinities between bacterial and mammalian bilayers which plays a vital role in designing less toxic and more active antimicrobial lipopeptides 65-71.
Drug target: A key biomolecule that is involved in a particular signaling and metabolic pathway associated with a specific disease or infectivity, pathology or survival of pathogenic microbe is called a biomolecular target (mostly a protein or nucleic acid). Antagonists, receptor agonists, modulators or inverse agonists, enzyme inhibitors or activators, ion channel blockers or openers are small molecules (drugs) that are complementary to the binding site of the target 72. Drug targets are not necessarily associated to cause disease but must be disease modifying 73. In most cases, small molecules are designed to inhibit or enhance the function of the target in their specific disease-causing pathway. Designed small molecules are complementary to the binding site of the target 72. Molecules are designed so as to improve specificity towards the target only and not to affect any other off-target molecule (anti-targets) because the interaction of drug with off-target may lead to undesired side effects 74. Similarity in the binding site, it may lead to cross-reactivity between closely related targets identified through homology of the protein sequence may have high risk of side effects 75.
Rational drug discovery: The searching for potential compounds complementary to the target protein starts with screening of libraries of drug compounds. This is generally done using screening assay in the wet lab experiments while a virtual screening is performed through computational tools if the structure of the target protein is available. In ideal condition, the selection of candidate drugs is based upon its drug-likeness, such as it should possess properties predicted in terms of oral bioavailability, minimal toxicity and appropriate metabolic and chemical stability 76,77. Lipinski’s rule of five, lipophilic efficiency and various scoring methods have been proposed in scientific literature to estimate drug metabolism 78,79. A large number of drug properties need to be optimized simultaneously throughout the drug design process; this can be achieved through the multi-objective optimization 80. Due to the limitations in current techniques of prediction of efficacy and selectivity, drug design is still bounded to rationality and serendipity 81. With the discovery of large molecules, the challenge becomes more obvious but with huge opportunities, however, it is still possible with advanced techniques if combined in accordance such as machine learning and artificial intelligence with advanced drug discovery methods like molecular dynamics simulation and QSAR 82.
Analogue-based drug design: "Analogy" term is derived from the Latin and Greek word analogia. Since 1791, it is in practice in natural science to describe functional and structural similarity 83. The definition is employed to the analogue of existing compound which shares therapeutic and clinical similarity with the parent compound. Simple traditional methods of medicinal chemistry are used to generate analogues of drug such as for the synthesis of homologues, isosteres, vinylogues, positional isomers, ring system transformation, synthesis of twin ring and optical isomers 39,79. Hence, the analogue-based approach is available with abundant opportunities for the designing of a large molecule. The lipopeptide molecule with the presence of a fatty acid chain of varied carbon length and a cyclic peptide moiety provide an enormous opportunity to tweak the structure to generate various analogous compounds from the parent microbial lipopeptide of natural origin by using a semi-synthetic approach.
Molecular docking: Computational molecular docking methods have been immensely used for designing and development of small-molecule therapeutics with greater success. Similar attempts have been done for the peptide therapeutics. Though the docking methods used for depicting the interaction of small molecules are not justified to apply to the larger and flexible peptide compounds. Hence, this brought the interest in the development of methods for protein-peptide docking which is greatly applied to the drug design and development process for large molecules. The protein-peptide docking methods are extensively discussed in the review article provided by Ciemny M et al 80. The major challenges of protein-peptide docking have been identified as the flexibility issue, a scoring problem that entails the selection of high accuracy models out of the generated various conformations and integrative modeling for integrating computational prediction into the experimental data. Various approaches to combat the hindrances and improve the accuracy level of predicting the interaction have been attempted to resolve 80. Recent development in protein-protein interaction and peptide docking has triggered the rapid development of computational approaches. So far, various drug discovery applications to handle peptide ligands have been designed which include virtual screening of inhibitors, predicting models of subangstrom-quality, prediction of specificity, experimental data interpretation and designing of peptides interfering with protein-protein interactions 80-82. Figure 4 is depicting the level of complexity in the drug discovery process for large molecule therapeutics.
Recent studies have suggested the utilization of low-Root-Mean Square Deviation (RMSD) models over commonly used RMSD which could provide better quality information on complex molecules which identifies and includes most of the key interactions. In the current scenario, molecular docking studies are supported by experimental studies and the ambiguous experimental data are also needed to be supported by computational studies to define key interactions, visualization of the receptor-inhibitor complex. It is expected that applications of protein-peptide docking will be explored with advanced techniques for scoring methods, flexibility modeling and integrative modeling 80,83. Protein therapeutics with the complexity in utilizing as drugs and synthetic peptides for the issues with chemical design, microbial lipopeptide with the semi-synthetic approach for drug designing of novel therapeutics, reserves a sweet spot amongst the two.
Molecular dynamics simulation: Molecular dynamics simulation is a computational program for analyzing the dynamics and physical interaction at the molecular and atomic level. The role of atomistic computational simulation in drug discovery is relatable to the experimental results. The approach is capable to provide more obvious and deeper insight into the interaction of target protein with a ligand 84. This includes the identification of allosteric and cryptic binding sites 85. This is more unbiased approach as compared to the traditional docking method wherein the interaction is rigid while simulation entertains the dynamics of participating molecules and the surrounding 86 (Figure 5). Hence this provides the validation of the docking study for interaction. It includes enhanced virtual-screening methods with the direct prediction of binding energy 87.
Computer simulation methodology has also been successfully used for exploring the mechanism of action of certain drugs including large molecule inhibitors such as peptides and lipopeptides and the effect of mutation and drug resistance studies on target protein are of help in understanding the drug resistance 88,89. The technique is also used for the prediction of specificity and selectivity towards the biological membranes. Molecular dynamics simulation study on lipo-peptide fengycin was done to explore their higher selectivity for fungal membrane than bacterial membrane, hence the drug is majorly used as an antifungal compound 89. In the current scenario simulation methods incurred with limitations in an approximation of required molecular forces and high computational cost. The future of computer-aided drug designing seems promising with the advancements of designed algorithms and power of computer facilities, wherein molecular dynamics simulation plays an important role 87.
Conclusion :
The drugs are majorly divided into two categories i.e., small molecule drugs used traditionally with oral bioavailability and molecular weight of less than 500 Dalton and large molecule biologics with molecular weight less than 5000 Dalton, which are not suitable for oral bioavailability and mostly delivered through injection. The small molecule inhibitors lack in reduced target selectivity due to the small size, hence off-targeting manifests in other side effects. Whereas, peptide therapeutics come with increased specificity for their targets due to multiple interactions. However, there is a need to explore such new categories of drugs and with reinvestigation on pharmaceutical properties of drugs with optimization of lead compounds for their bioavailability, metabolic instability and membrane permeability 9,90.
This review is more focused on natural peptides such as lipopeptides of microbial origin which are composed of a hydrophobic (fatty acid moiety) and hydrophilic cyclic peptide core, which itself avails the multiple features of structural rigidity, membrane permeability, target specificity and membrane selectivity 63. Rather the research on synthetic peptides includes exhaustive lab work, which comes with multiple series of reactions to generate such complex therapeutics. The utilization of large molecules in computer-aided drug discovery is explored less due to the complex structures of such compounds like lipopeptide with cyclic peptide core with hexa to deca fatty acid side chain. But various drug discovery platforms are available to handle such complex molecules. Schrodinger LLC. Provide Maestro platform to perform molecular docking with extra precision mode and specifically designed application for peptide docking. Various free web servers such as Autodock Vina on Pyrx platform, DINC to perform incremented docking, fragmented docking, peptide docking servers such as PeptiDock, Z-Dock, M-Dock, and Rosie: TheRosetta docking application are compatible to handle docking studies of large molecules. Databases have also been available to access large molecules such as BioSurfDB, PepBank, THPdb, LppDB, Peptide Atlas, Antimicrobial peptide database etc.
Computational approach with peptides of natural origin brings the opportunity to screen and optimize the drug molecule for the desired pharmaceutical properties; though the advance computational methods must be coupled together to produce more obvious outcomes. This includes structural dynamics study of the interaction of drug molecule on target for a timescale manner and identification of principal components of the interaction. This gives the deeper insight to modify the drug through analogue-based drug discovery. Such studies are further explored towards a semi-synthesis approach of drug discovery; wherein whole molecule synthesis is replaced with just certain desired modification on the parent molecule to generate a more promising candidate drug. The article is composed of potential opportunities and challenges incurred in the field of drug discovery of large molecules.
Acknowledgement :
We are thankful to the National Institute of Technology Raipur and Chhattisgarh Council of Science and Technology (CCOST) (Project number 2487/ CCOST/MRP/2016, Raipur dated 25.01.2016), India for providing the necessary facilities to prepare the manuscript and permission to publish it.
Conflict of Interest :
Authors declare no conflict of interest.
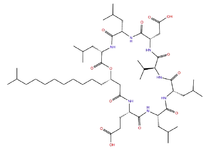
Figure 1. 2Dimesional structure of lipopeptide Surfactin (cyclic peptide with fatty acid side chain).
|
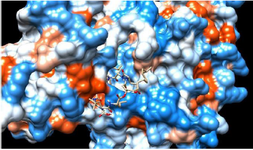
Figure 2. Molecular docking interaction: cyclic peptide drug docked with target protein (electrostatic surface interaction). The ability of cyclic peptide to inhibit target from multiple sites.
|
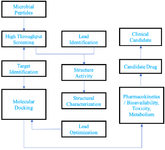
Figure 3. Generalized protocol: computer-aided drug designing (workflow for computer-aided drug discovery of peptides derived from microbial origin).
|
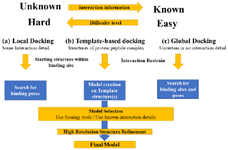
Figure 4. Level of complexity and types of molecular docking for protein-peptide docking.
|
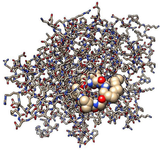
Figure 5. Molecular interaction of a cyclic peptide with target protein-Silhouette view (Efficient inhibition of the active site): Ball-stick model for target protein docked by lipopeptide ligand in sphere form.
|

Table 1. Large molecule therapeutics with mechanism of action and current status
|
|