An in silico Design, Expression and Purification of a Chimeric Protein as an Immunogen Candidate Consisting of IpaD, StxB, and TolC Proteins from Shigella spp.
-
Fathi, Javad
-
Department of Bacteriology and Virology, Faculty of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
-
Student Research Committee, Faculty of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
-
 Nazarian, Shahram
Department of Biological Sciences, Faculty of Science, Imam Hossein University, Teh-ran, Iran, nahalhadi@gmail.com
Nazarian, Shahram
Department of Biological Sciences, Faculty of Science, Imam Hossein University, Teh-ran, Iran, nahalhadi@gmail.com
-
Kordbacheh, Emad
-
Department of Biological Sciences, Faculty of Science, Imam Hossein University, Tehran, Iran
-
Hadi, Nahal
-
Department of Bacteriology and Virology, Faculty of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
Abstract: Background: Shigella spp. is the cause of dysentery and is widespread worldwide. On the other hand, antibiotic resistance is increasing in this bacterium. Bioinformatics is a new approach to vaccine and drug design involving the selection of appropriate antigens. This study aimed to design a chimeric protein consisting of IpaD, StxB, and TolC proteins from Shigella through a bioinformatics approach as an immunogen candidate.
Methods: The sequences of ipaD, stxB, and tolC genes were obtained. Additionally, the immunogenic regions of the associated protein, physicochemical characteristics, protein structures, B and T cells epitopes, and molecular docking were determined using in silico servers. Besides, the chimeric gene was synthesized following sequence optimization by utilizing the codon usage of Escherichia coli (E. coli). The expression of the recombinant protein was confirmed via SDS-PAGE and Western blot technique.
Results: The residues 41-160 of IpaD, 21-89 of StxB, and 40-335 of TolC were selected. According to half-life, instability, and buried indices, IpaD-StxB-TolC was selected as the best arrangement. The Ramachandran plot showed that 97.077% of the amino acids were in the favored area. Linear and conformational epitopes were also present throughout the chimeric protein sequence. Moreover, the C-ImmSim server indicated that IgG and IgM titers could reach desirable values by the third injection. Furthermore, the stability of the mRNA-optimized gene was enhanced, increasing the Codon Adaptive Index (CAI) to 0.9. Finally, the chimeric gene was transferred to E. coli BL21, and the expression of the 60.6 kDa recombinant protein was confirmed.
Conclusion: The results indicated that the recombinant protein could act as a proper immunogen candidate against Shigella spp.
Introduction :
Diarrheal diseases are the leading cause of death amongst children in developing countries. These diseases are also the second leading cause of death worldwide after respiratory diseases. The Enterobacteriaceae family is one of the most important causes of these diseases 1,2. Shigella is one of the main causes of gastrointestinal diseases and diarrhea in this family and includes four species, namely Shigella boydii, Shigella dysenteriae, Shigella flexneri (S. flexneri), and Shigella sonnei. Diarrhea caused by Shigella can be a simple watery or dysentery (bloody) diarrhea. Diarrhea caused by Shigella is also known as Shigellosis and is mainly common in developing countries with poor health and dense populations 3,4. According to the latest studies, 80 million dysentery cases and more than 700,000 associated deaths are reported annually 5,6.
Shigella contains various virulence factors (binding and invasion factors) and toxins, which are considered pathogens causing infection and death 7. Among its invasion factors, TolC protein plays an important role in binding of bacteria to host cells, secretion of bacterial toxins, and excretion of antibiotics from bacteria. This protein can create a water-filled channel across the inner and outer membranes, leading molecules to secret from the bacteria without entering the periplasmic space. Therefore, the TolC plays a critical role in increasing multidrug resistance and is important for the survival of pathogenic bacteria during infection 8,9. Another virulence factor contains Invasive plasmid antigens (Ipa) that enter the host cell through the Type III Secretion System (T3SS) 10. These proteins include IpaA, IpaB, IpaC, and IpaD, the last of which controls T3SS secretion, phagosome escape, and macrophage apoptosis. The IpaD is located at the tip of the T3SS apparatus by IpaB, which leads to the bacteria-host cell binding and invading. Due to the IpaD location at the tip of the T3SS and its conservation among Shigella serotypes, it has been recognized as a candidate antigen for inducing immune system response 11,12. Following the binding and colonization of bacteria in the intestine, Shiga toxin (Stx) is produced. This toxin inhibits protein synthesis, thus causing ulcers in intestinal cells 13. The Stx consists of two subunits, A and B. The A subunit is linked non-covalently to five identical B subunits. The role of the subunit B is to bind, create pores, and enter endothelial cells, while the subunit A has an enzymatic function and leads to the inhibition of protein synthesis and apoptosis in host cells 14,15.
Antibiotics are one of the important strategies for preventing the spread and treatment of Shigellosis, leading to the improvement of health conditions 16. However, due to the increasing and irregular use of antibiotics, antibiotic resistance is emerging. In the recent decades, the production of ESBL and AmpC beta-lactamases in Shigella spp. has increased worldwide. Hence, the production of various forms of these enzymes can be considered the main mechanism of resistance against beta-lactam agents; i.e., cephalosporins. This mechanism can reduce the efficacy of treatment strategies used to cure Shigellosis 17. In addition, various strategies have been used for decades for making safe and effective vaccines against Shigella spp., but a licensed, safe, and well-functioning vaccine is not available yet. One of the novel ways to produce a vaccine against Shigella is chimeric protein vaccines. These polyvalent vaccines can reduce the cost of producing and injecting vaccines 18,19. Chimeric proteins carrying effective epitopes or adjuvant sequences increase the possibility of eliciting a broad cellular or humoral immune response 20,21.
Today, a new field of science so-called bioinformatics has been developed with the evolution of computer science and information technology and the ability to analyze biological data as well as the sequencing of genomes and proteins 22-28. Immunoinformatic approaches can be used in various fields including prediction of B and T cell epitopes, immune response simulations, antigen receptor interaction, etc. Utilization of computational methods can reduce the time and cost of experimental phase in various medical-diagnostic fields 29. Therefore, the present study aims to design a chimeric protein consisting of IpaD, StxB, and TolC proteins from Shigella spp. through a bioinformatics approach as an immunogen candidate.
Materials and Methods :
Retrieval of antigen and cassette design
Initially, the amino acid sequences of StxB (UniProt entry: Q7BQ98), IpaD (UniProt entry: P18013), and TolC (UniProt entry: P0AAX2) proteins were obtained from an online database (www.uniprot.org) and were saved in FASTA format. Then, to determine the conserved domains and antigenic segment, the Clustal Omega server (https://www.ebi.ac.uk/Tools/msa/clus-talo/) was used as provided by the EBI database (https: //www.ebi.ac.uk/Tools/msa/clustalo/). The related sequences for designing the chimeric construct were StxB (accession No. EF685161.1), IpaD (accession No. GQ201921.1), and TolC (accession No. X00016). To maintain the structural stability of the chimeric protein and non-interference of domains, rigid linkers (EAAA-K) including glutamic acid, alanine, and lysine were applied. The 6-XHis sequence was also embedded to the C-terminal of the recombinant protein to achieve protein purification and identification.
The physicochemical characteristics of the construct
In this step, construct properties were evaluated using the ExPASy ProtParam tool (http://us.expasy.org/tools/ protparam.html). These features included the physicochemical properties, molecular weight, theoretical isoelectric point (pI), instability index, extinction coefficient, half-life, aliphatic index, Grand Average Hydropathy (GRAVY), and the total number of positive and negative residues 30.
Prediction of the secondary structure
PHD (http://npsa-pbil.ibcp.fr/cgi-bin/npsa) 31, PORTER (http://distill.ucd.ie/porter/) 32, GORV (http://gor. bb.iastate.edu/) 33, SOPMA (http://npsapbil. ibcp.fr/cgi-bin/npsa) 34, and PREDATOR (http://npsa-pbil.ibcp.fr/ cgi-bin/npsa) 35 online servers were used to determine and predict the second structure.
Prediction of the 3D structure
I-TASSER server (http://zhanglab.ccmb.med.umi-ch.edu/I-TASSER/) was used to predict the three-di-mensional structure and to determine the best order of the StxB, IpaD, and TolC sections in the chimeric protein. Finally, the predicted PDB file was used as the conformational structures for further evaluations 36.
Tertiary structure refinement
Using the UCSF Chimera v1.15, the best predicted model for chimera was viewed and assessed for topology errors 37. Afterwards, the 3Drefine and GalaxyWEB servers (http://galaxy.seoklab. org/cgi-bin/sub-mit.cgi?type=REFINE) were used to refine the best obtained model 38,39. Atomic-level energy minimization and optimization of the hydrogen bonding network so-called MESHI force fields were performed by the i3Drefine refinement algorithm. The GalaxyRefine server was also used to promote the quality of local and global structures via side-chain repacking using the molecular dynamics simulation method.
Validation of the 3D structure model
In order to appraise the progress of crystallographic model building and refining, ERRAT server (http:// services.mbi.ucla.edu/ERRAT/) was used as a protein structure authentication algorithm. Ramachandran plot and ProSA web server were also utilized for further confirmation of the predicted 3D structure. Finally, NetSurfP server (http://www.cbs.dtu.dk/services/Net-SurfP/) and Oklahoma University web-based service (http://www.biotech.ou.edu/) were used to determine the surface residues in the recombinant protein as well as the protein solubility according to the final produced and refined model.
Validation of structural quality
Some indices like Root Mean Square Deviation (RMSD) and TM-score were employed to verify the 3D morphology of the predicted models. They were presented by the predictor servers. Furthermore, to distinguish the errors of the best predicted model, its 3D format (PDB) was uploaded in ProSA and SAVES v5.0 web services 40,41. The quality of the stereochemistry results was also investigated in the Ramachandran plot in the RAMPAGE server 42.
Protein structures
Firstly, the amino acid composition was analyzed using the protein predictor server for disulfide bonds 43. The GOR and PHD methods were utilized for homology modeling of the secondary structure consisting of alpha helices and beta sheets. Hence, the PSIPRED and scratch protein predictor servers were operated in these terms as well as in the prediction of solvent accessibility 44,45. For generating a validated tertiary structure, ab initio and comparative modeling algorithms are customary. Based on experience, the RaptorX (http://rap-torx.uchicago.edu/StructurePropertyPred/predict/) and the Iterative Threading Assembly Refinement (I-TASSER) servers (https://zhanggroup.org/I-TASSER/) are more reliable. Thus, they were exploited 46,47. Finally, the DOG 1.0 program was used to visualize the produced 3D models 48.
Molecular docking
Molecular docking is a good approach to drug design. According to the literature, the Stx has an interaction with the Gb3 protein receptor. Hence, the best crystal structures with the lowest resolution of the Gb3 receptor were retrieved from the RCSB database (A.n 1D1K), and docking was done following the removal of H2O, heteroatoms, and unrelated residues and the addition of polar hydrogens. In addition, the structure of the receptor protein was prepared with the help of the PyMOL software. Moreover, ClusPro v2.0 and HADDOCK servers were used to simulate the receptor‐ligand docking 49,50. The ClusPro can rotate ligand and receptor proteins around each other. This program performs a rigid docking by utilizing Fast Fourier Transform (FFT) methods named PIPER. Subsequently, the server scores probable docked structures with the lowest energy and stable cluster size based on the Semi-Definite programming-based Underestimation (SDU) algorithm and Monte-Carlo simulation refinement. In the present study, High Ambiguity Driven protein-protein Docking (HADDOCK) 2.4 server was employed using a flexible docking approach. HADDOCK uses a group of python scripts that use Nuclear Magnetic Resonance (NMR) and crystallography data plus ab-initio docking. This server performs modeling based on Optimized Potentials for Liquid Simulations (OPLS) force field in three steps; i.e., rigid-body energy minimization, semi-flexible refinement, and final refinement. Then, best docking proteins were submitted to the PRODIGY server (PROtein binDIng enerGY prediction) to obtain free energy (ΔG) and dissociation constant (Kd) data and were finally visualized in the PyMOL v.2.3 software 51. After docking, the complex structure of the designed candidate vaccine with the Gb3 receptor was analyzed by iMOD and CABSflex servers to determine the stability and mobility of the complexes.
B and T cells epitopes
At this stage, B-cell linear epitopes with various lengths were investigated using the Bcepred server 52. Non-linear B-cell epitopes were also evaluated using CBTOPE, which could predict conformational epitopes by the first protein structure or sequence 53. The residues overlap was considered using the IEDB server to determine the T cell epitope competence 54. For the chosen peptides, Cytotoxic T Lymphocytes (CTL) and Helper T Lymphocytes (HTL) epitopes were identified. Therefore, Major Histocompatibility Complex (MHC)-I and MHC-II epitopes were predicted for human alleles. Finally, the full Human Leukocyte Antigen (HLA), the best supertypes of MHC-I, and a high overall score for MHC-II peptides were chosen. According to the IEDB recommended value with a word length of 16 residues, the binding affinity of the MHC molecules and the half-maximal inhibitory concentration (IC50) cut-off of the HTL epitopes were set.
Sequence optimization and mRNA structure prediction
RNA Predict Secondary Structure Server and Vienna RNA Web Servers (RNAfold) were used to investigate the chimeric gene parameters as well as the mRNA secondary structure 55,56. The sequence optimization was performed for high-level expression in Escherichia coli (E. coli). The RNA secondary structure was compared before and after codon optimization. The minimum energies of the native and optimized mRNA were compared, as well 57.
Immune simulation
C-ImmSim server (https://www.iac.rm.cnr.it/~filip-po/projects/c-immsim-online.html) was used to predict the in silico immune response profile of the chimeric protein. This server could describe the humoral and cellular immune responses against the recombinant protein. Finally, three administrations were set with four-week intervals.
Expression, verification, and purification of the recombinant chimeric protein
After designing and ordering the chimeric IpaD-StxB-TolC gene in pET28a (ShineGene, China), this structure was transferred to E. coli BL21 (DE3) by the heat shock method. Then, by adding 1 mM isopropyl-β-d-thiogalactoside (IPTG; Sigma, USA) to the bacterial culture medium, recombinant protein production was induced and incubated at 37°C for 5 hr. After that, the culture medium was centrifuged (5000×g at 25°C for 10 min) and the bacterial precipitate was mixed with the lysis buffer (100 mM NaH2PO4, 10 mM Tris-HCl, 8 M urea, pH=8.0). In the next step, after sonication (6 times for 10 s with high power), the lysed cell was centrifuged at 10000 g at 4°C for 15 min, the precipitate was discarded, and the supernatant was loaded in the Ni-NTA chromatography column (Qiagen). Protein elution was achieved using the elution buffer containing 100 mM NaH2PO4, 10 mM Tris-base, and 8 M urea (pH=4.2). SDS PAGE 10% and Western blot analysis using anti-His tag antibody were performed to confirm the recombinant protein expression.
Results :
Sequence arrangement and physicochemical characteristics of the construct
StxB, IpaD, and TolC proteins were selected as acceptable immunogens and immunogen candidates based on the previous studies. Protein sequences of StxB, IpaD, and TolC were retrieved from the Uniprot database and were aligned with the Clustal omega server. To design the recombinant protein, residues 41-160 of IpaD, 21-89 of StxB (without the signal peptide), and 40-335 of TolC were selected. Three repeats of EAAAK (as a rigid linker) were also placed among the recombinant protein subunits to maintain the structure and independent function of each protein subunit (Figure 1). Finally, in order to achieve protein purification and identification, the 6-XHis sequence was embedded to the recombinant proteins C-terminal. For the best arrangement, several factors including half-life, instability index (https://web.expasy.org/protparam/), VaxiJen score (http://www.ddg-pharmfac.net/vaxijen/ VaxiJen/VaxiJen.html), solvent access (http://raptorx. uchicago.edu/StructurePropertyPred/predict/), protein solubility (https://proteinsol.manchester. ac.uk/), and allergencity evidence (https://www.ddgpharmfac.net/ AllerTOP/index.html) were evaluated (Table 1). According to half-life, instability, and buried indices, IpaD-StxB-TolC was selected as the best arrangement for the desired cassette.
Prediction of the structures
The second structure was evaluated using GORIV, and the third structure (Figure 2) was built via I-TA SSER, RaptorX, and Phyre2 model (Table 2). After that, the most morphologically favorable and stable models were selected. The quality of the predicted structure was evaluated using the Ramachandran plot and the PROSA software (Figure 3).
Molecular docking and dynamic simulations
The docking of the structure of the designed candidate vaccine with Gb3 by the ClusPro server led to the generation of 10 models. The flexible dock was predicted by the HADDOCK server. The receptor protein had interactions with the Stx regions of the protein. Regarding ClusPro, based on the lowest energy scores (-1042.2 kJ/mol), model number 0 was selected for the Gb3-candidate vaccine complex as the best-docked complex with ΔG of -18.3 (kcal mol-1) by PRODIGY. The HADDOCK server predicted 81 structures in 15 clusters, with the best cluster presenting a score of 97.5±8.0 and ΔG of -16.6 (kcal mol-1) at 37.0°C. Moreover, the LigPlot analysis indicated that the receptor-protein complex included salt bridges, hydrogen bonds, and non-bonded contacts (Figure 4A). CABS-flex and iMOD servers performed the molecular dynamic simulation of the protein receptor interaction. As shown in figure 4B, B-factor mode presentation of the complex showed less deviation in atomic fluctuation. Consequently, it was a solid interaction. Besides, the eigenvalue of the protein-receptor was 4.577188e-6, which implied its stability.
Prediction of B cell epitope
Vaccine candidates should be able to induce the response of both B and T cells. Determination of B linear epitopes is possible using their innate affinity for amino acids and bioinformatics algorithms. The linear epitopes of B cells in the chimeric protein predicted by the BCpred server have been depicted in figure 5. Accordingly, the linear epitopes were present throughout the chimeric protein sequence and belonged to three subunits (IpaD-StxB-TolC) of this protein. The results of predicting the conformational B-cell epitopes using CBTOPE and Discotope servers have been presented in table 3. Based on the results, conformational B-cell epitopes were present throughout the recombinant pro tein sequence and belonged to the three abovementioned proteins.
Prediction of T cell epitopes
The MHC-I and MHC-II binding prediction tools from the IEDB server were employed to investigate T-cell epitopes (Table 4). According to the results, five most frequent alleles in eight residue lengths were chosen for MHC class I. Additionally, five most frequent alleles in 14 residue lengths were selected for MHC-II.
Immune simulation
Generally, the C-ImmSim can be successfully used to determine immune responses. As illustrated in figure 6, recognition of the subunit protein and the proper immune response resulted in an increase in antibody titer. In the second injection, the IgG and IgM immunoglobulin titers started to increase, reaching the maximum values in the third injection.
Sequence optimization and mRNA structure prediction
To enhance the transcriptional and translational function of E. coli, the sequence encoding the IpaD-StxB-TolC chimeric gene was optimized via the optimizer and the rare codon analysis tool. As shown in table 5, the Codon Adaptation Index (CAI) was 0.74 before optimization and 0.90 after that (ideal value= 0.8-1.0). The GC content also increased from 46.45% to 54.63%) ideal value=30-70%).
The thermodynamic properties of the secondary structure of mRNA indicated an increase in the stability of the mRNA optimized gene. The RNA secondary structure of the chimeric gene was predicted using the Mofld algorithm (Figure 7). The predicted structure did not have stable hairpin and pseudoknot at the 5' site of the mRNA.
Expression, purification, and verification of the chimeric protein
The recombinant vector was transferred to the E. coli BL21 strain, inducing recombinant protein expression by IPTG. A 60/6 kDa protein band on SDS-PAGE 10% showed a high level of recombinant protein expression (Figure 8A). Purification of the protein was carried out by Ni-NTA chromatography column and under native conditions. SDS-PAGE 10% analysis revealed the presence of the recombinant protein in the eluted fraction (IM 250) (Figure 8B). Finally, the recombinant protein was confirmed by western blot using an anti-His tag antibody (Figure 9).
Discussion :
Shigella spp. are intracellular pathogenic bacteria that colonize the gastrointestinal tract, especially intestinal cells, and cause bacillary dysentery by invading these cells. Bacillary dysentery has been a problem in developing countries, mainly causing symptoms and diseases in children, immunocompromised patients, and elderly individuals 58. Invasion and pathogenicity of Shigella are increased through the secretion system, binding, invasion factors, bacterial toxins, and excretion of antibiotics from bacteria. Shigella, as an intra-cellular pathogen, has challenged antibiotic treatment approaches. Although fluoroquinolones, cephalosporins, and azithromycin are used to treat Shigella, resistance to these antibiotics is growing, which is a major concern in treating this bacterium 59. Designing effective vaccines against Shigella can be a good way to overcome antibiotic resistance as well as to control and prevent the spread of this bacterium 60. Although most live vaccines can provide better protection against pathogens compared to other vaccines, the attenuation process and achievement of minimum toxicity is a difficult process. On the other hand, due to the induction of immunosuppressive conditions, the use of live Shigella vaccines can interfere with the immune response of the host and cause immune reactions 61,62. Recombinant and subunit vaccines are a new and effective strategy against pathogens such as Shigella. The use of several different antigens in a chimeric protein can induce and produce several antibodies simultaneously against the pathogen 63. IpaD is a highly conserved protein among Shigella serotypes and is located at the tip of the T3SS. The role of this protein is to bind and invade bacteria to host cells 11. Previous studies have proved that the IpaD has the ability to stimulate the immune system and produce antibodies. Thus, any
mutation or deletion in this protein leads to defects in bacterial functions including secretion control and insertion of translocators into host cell membranes 12,64. S. dysentery secretes Stx toxin, which binds to host ribosomes and results in the apoptotic death of host cells by inhibiting protein synthesis 13,65. The StxB subunit binds to the host cell, while the StxA subunit plays an enzymatic role and inhibits protein synthesis. The previous studies have also emphasized that StxB has an adjuvant role in addition to the ability to induce antibody production 14,66,67. Furthermore, numerous studies have reported that the outer membrane channel protein (TolC) is involved in multidrug resistance in gram-negative bacteria including S. flexneri. TolC is also involved in the binding of bacteria to host cells, secretion of bacterial toxins, and excretion of antibiotics from bacteria 8,68.
Nowadays, bioinformatics tools and approaches can help design chimeric proteins and vaccine candidates. Thus, online bioinformatics tools were utilized in the current investigation to design a vaccine candidate against Shigella spp. based on B and T cells epitopes and production of antibodies (IgG and IgM). Considering the increasing antibiotic resistance, lack of an effective vaccine against Shigella spp., and advantages of bioinformatics, this study aimed to design a chimeric protein consisting of IpaD, StxB, and TolC proteins from Shigella through a bioinformatics approach as an immunogen candidate. In this study, the protein se-quences of IpaD, StxB, and TolC were retrieved from the Uniprot database and were aligned with the Clustal omega server. Residues 41-160 of IpaD, 21-89 of StxB (without the signal peptide), and 40-335 of TolC were selected. Based on the researchers’ past experience, Kordbacheh et al and Felegary et al the selected regions were fused via the EAAAK linker and a novel construct was built 69,70. The EAAAK rigid linker is appropriate for bifunctional chimeric protein. Based on half-life, instability, and buried indices, IpaD-StxB-TolC was found to be the best arrangement. The molecular weight of the chimeric protein was 60.6 kDa. The second structure was evaluated via GORIV, and the third structure was built based on the RaptorX model. Accordingly, Helix 61%, strands 11%, coils 26%, and the lack of a major burial domain (18%) were determined. The structure quality was evaluated by the Ramachandran plot and the PROSA software. Based on the findings, 97.077% of the amino acids were in the favored area. Additionally, the Z-score plots determined X-ray and NMR structures and confirmed the angles and distances of the residues. Docking of the vaccine candidate with Gb3 (receptor of StxB) led to the generation of 10 models. Model number 0 with lowest energy scores (-1042.2 kJ/mol) was selected for the Gb3-candidate vaccine complex as the best-docked complex with ΔG of -18.3 (kcal mol-1). Moreover, the HADDOCK server predicted 81 structures in 15 clusters, with the best cluster showing a score of 97.5±8.0 and ΔG of -16.6 (kcal mol-1) at 37.0°C. Besides, the receptor-protein complex included salt bridges, hydrogen bonds, and non-bonded contacts. Molecular dynamic simulation of the protein receptor interaction indicated that the B-factor mode presentation of the complex had less deviation in atomic fluctuation. Consequently, it was a solid interaction. The eigenvalue of the protein-receptor was also 4.577188e-6, implying its stability.
Since vaccines should be able to maximally stimulate the immune system especially B and T cells, and determination of epitopes has a fundamental role in vaccine design, the BCpred server was used to determine the linear B-cell epitopes and CBTOPE and Discotope servers were utilized for predicting the conformational B-cell epitopes. The results revealed that linear and conformational B-cell epitopes were present throughout the chimeric protein sequence and belonged to three subunits (IpaD-StxB-TolC). Furthermore, the IEDB server was used for the prediction of T cell epitopes, which showed five alleles in eight residue lengths for MHC-I and five alleles in 14 residue lengths for MHC-II. Assessment of the immune responses and production of antibodies against the chimeric protein through C-ImmSim indicated that the highest IgG and IgM titers were obtained after the third injection. After optimizing the sequence encoding the chimeric protein, CAI and GC content increased to 0.90 and 54.63%, respectively and led to an increase in transcription and translation in E. coli. The stability of the mRNA optimized gene was enhanced, as well. Additionally, the predicted mRNA structure had no long stable hairpin and pseudoknot at the 5' site. In the last step, after bioinformatics studies and preparation of the recombinant vector, this vector was transferred to E. coli BL21 strain. After inducing the bacteria for the expression of the recombinant protein, protein expression was assessed by the SDS-PAGE method and protein confirmation was performed via western blot using anti-His tag antibody. These two methods confirmed the presence of the chimeric protein with the molecular weight of 60.6 kDa. In the continuation of this study, this recombinant protein can be evaluated for antibody production and immunogenicity both in vitro and in vivo.
Conclusion :
Shigella spp. is emerging as a global problem due to deadly diseases as well as increased antibiotic resistance. In this context, the lack of an effective vaccine against Shigella is a red alert for public health. Today, the use of bioinformatics software has led to more effective and faster studies in vaccine development. In the current research, toxin, T3SS, and binding factors of Shigella spp., as a vaccine candidate, were evaluated with the help of bioinformatics software. The findings indicated that the chimeric protein containing IpaD-StxB-TolC had a high ability to stimulate the immune system and, as a vaccine candidate, could lead to immunogenicity and antibody production.
Acknowledgement :
The authors would like to thank Ms. A. Keivanshekouh at the Research Consultation Center (RCC) of Shiraz University of Medical Sciences for improving the use of English in the manuscript.
Conflict of Interest :
The authors declare no conflict of interests.
Funding: This work was supported by Shiraz University of Medical Sciences, Shiraz, Iran (IR.SUMS. REC.1400.347). It was done based on the results of the PhD dissertation written by Javad Fathi under the supervision of Dr. Nahal Hadi and Dr. Shahram Nazarian.
Data availability statement: The data are available from the corresponding author upon request.
Ethics of Approval Statement :
Not applicable.
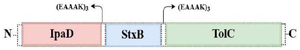
Figure 1. Schematic presentation of the recombinant protein construct consisting of StxB, IpaD, and TolC proteins bound together by the EAAAK linkers.
|
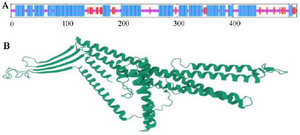
Figure 2. Graphical presentation and assessment of the secondary structure (A). Cartooned secondary structure showing helix 61%, strands 11%, coils 26%, and 50% exposure. Concerning the tertiary structure, the RaptorX predicted the lack of a major burial domain (18%), as shown by the UCSF Chimera (B).
|
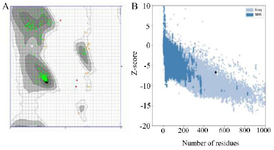
Figure 3. As the Ramachandran plot views the error values of the residues, 97.077% of the amino acids were in the favored area (A). Z-score plots of the ProSA server compared the subunit protein to the pre-determined X-ray and NMR structures and confirmed the angle and distance of the residues (B).
|
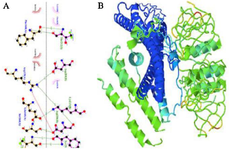
Figure 4. Molecular docking and dynamic simulations. A) the receptor-protein complex, B) the molecular dynamic simulation of the protein receptor interaction.
|
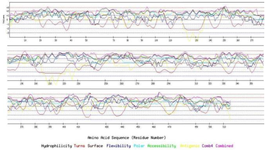
Figure 5. B-cell epitopes from IpaD-StxB-TolC full-length proteins using the BCpred server.
|

Figure 6. The C-ImmSim immune response simulation immunoglobulins levels after the injections. The hypothetical administration of the vaccine was carried out in three injections given four weeks apart.
|
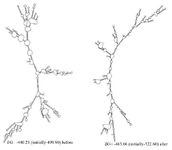
Figure 7. Prediction of the RNA secondary structure of the chimeric gene using the Mofld algorithm.
|
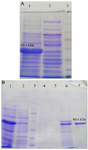
Figure 8. A) Expression of the recombinant protein. Lane 1: Expression of the recombinant protein induced by IPTG. Lane 2: Expression of the recombinant protein without IPTG. Lane 3: Protein weight marker (prestained protein ladder, 10-170 kDa). B) SDS-PAGE 10% of the chimeric protein (IpaD-StxB-TolC). Lane 1: Cell lysate E. coli containing pET28a induced by IPTG. Lane 2: Flow-through. Lane 3: Protein weight marker (prestained protein ladder, 10-170 kDa). Lane 4: Wash C fraction (pH=6.3). Lane 5: Wash D fraction (pH=5.9). Lane 6: Wash E fraction (pH=4.3). Lane 7: Elution fraction (IM 250).
|
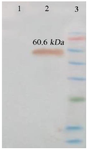
Figure 9. Identification of protein expression (IpaD-StxB-TolC) using western blot analysis. The anti-His antibody detected the expression of a chimeric protein (60.6 kDa). Lane 1: Control (Bovine Serum Albumin, BSA). Lane 2: Chimeric protein. Lane 3: Protein weight marker (prestained protein ladder, 10- 170 kDa).
|
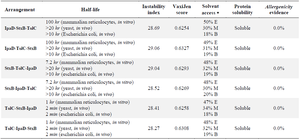
Table 1. Determining the best arrangement and the physicochemical characteristics
* E: Exposure, M: Mediate, B: Buried.
|
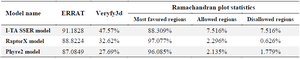
Table 2. The third structures predicted by different tools and comparison of their scores
|
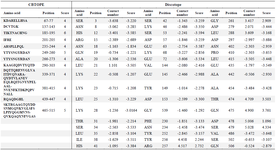
Table 3. Conformational B-cell epitopes from full-length IpaD-StxB-TolC protein using the CBTOPE and discotope servers
|
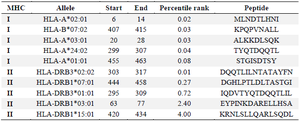
Table 4. Selection of peptides containing T cell epitopes from the chimeric protein by MHC-I and MHC-II binding prediction algorithms
|
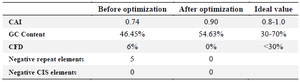
Table 5. Sequence optimization
|
|