Prioritizing Candidate Genes for Type 2 Diabetes Mellitus using Integrated Network and Pathway Analysis
-
Prakash, Tejaswini
-
Genetics and Genomics Lab, Department of Studies in Genetics and Genomics, University of Mysore, Manasagangothri, Mysuru – 570 006, Karnataka, India
-
 B Ramachandra, Nallur
Department of Genetics and Genomics, University of Mysore, Manasagangothri, Mysuru–570 006, Karnataka, India, nallurbr@gmail.com
B Ramachandra, Nallur
Department of Genetics and Genomics, University of Mysore, Manasagangothri, Mysuru–570 006, Karnataka, India, nallurbr@gmail.com
-
Genetics and Genomics Lab, Department of Studies in Genetics and Genomics, University of Mysore, Manasagangothri, Mysuru – 570 006, Karnataka, India
Abstract: Background: Type 2 Diabetes Mellitus (T2DM) has emerged as a major threat to global health that fosters life-threatening clinical complications, taking a huge toll on our society. More than 65 million Indians suffer from T2DM, making it one of the leading causes of death. T2DM and associated complications have to be constantly monitored and managed which reduces the overall quality of life and increases socioeconomic burden. Therefore, it is crucial to develop specific treatment and management strategies. In order to achieve this, it is essential to understand the underlying genetic causes and molecular mechanisms.
Methods: Integrated gene network and ontology analyses facilitate prioritization of plausible candidate genes for T2DM and also aid in understanding their mechanistic pathways. In this study, T2DM-associated genes were subjected to sequential interaction network and gene set enrichment analysis. High ranking network clusters were derived and their interrelation with pathways was assessed.
Results: About 23 significant candidate genes were prioritized from 615 T2DM-associ-ated genes which were overrepresented in pathways related to insulin resistance, type 2 diabetes, signaling cascades such as insulin receptor signaling pathway, PI3K signaling, IGFR signaling pathway, ERBB signaling pathway, MAPK signaling pathway and their regulatory mechanisms.
Conclusion: Of these, two tyrosine kinase receptor genes-EGFR and IGF1R were identified as common nodes and can be considered to be significant candidate genes in T2DM.
Introduction :
Type 2 Diabetes Mellitus (T2DM) is a major disorder of the metabolic and endocrine system which has gained the status of "global epidemic" owing to the increasing number of diagnosed cases worldwide 1. According to the 2019 report by International Diabetes Federation (IDF), 463 million people in the age group of 20-64 years were diagnosed with T2DM worldwide, which is approximately 8.8% of the adult population. This is further predicted to increase by 60-70% by 2030. In addition, 374 million individuals are said to be in pre-diabetic stage, displaying Impaired Glucose Tolerance (IGT). Each year, 3.7 million deaths occur as a consequence of T2DM and high blood glucose, making it the 8th leading cause of death. Globally, India ranks second with 77 million individuals diagnosed with T2DM. It is further predicted to increase to 101 million by 2030 and 134.2 million by 2045 2. Besides being an important health concern on its own, T2DM can also lead to severe complications of the vascular, renal and ophthalmic systems manifesting as Coronary Artery Diseases (CAD), Peripheral Artery Disease (PAD), diabetic nephropathy, diabetic retinopathy and diabetic neuropathy. In view of these debilitating consequences, immense efforts have been made towards understanding the molecular mechanisms of T2DM using high-throughput technologies, paving way for novel treatment and disease-management strategies. Currently, genome and transcriptome sequencing with bioinformatics analyses have been used widely to successfully identify numerous genes and functional pathways associated with pathophysiology of T2DM. However, defining the causal molecular players and their mechanism of action from the many identified gene sets is challenging and time consuming. In silico analysis using interaction network approach provides a practical solution to reduce noise and prioritize significant candidate genes for functional experiments.
System biology and protein interaction network approaches are being increasingly used to study complex diseases such as T2DM, CAD, Autism Spectrum Disorders (ASD) and cancers 3-8. The foundation for these approaches is the knowledge that protein molecules interact with each other to perform particular functions that are generally represented as interaction networks. Ding et al 9 used differential gene expression profile of pancreatic β-cells integrated with Protein‑Protein Interaction (PPI) network and functional analysis to identify HNF1A, STAT3, SERPING1, ANPEP and Glucocorticoid Receptor (GR) as crucial genes involved in the development of T2DM. Similarly, Lin et al 10 identified 36 differentially expressed genes between diabetic and non-diabetic pancreatic islets that were significantly enriched in PI3K-Akt signaling pathway, pathways in cancer, cytokine–cytokine receptor interaction and Rheumatoid arthritis. Furthermore, 10 important hub genes were established of which IL6, MMP3, MMP1 and IL11 were regarded potential biomarkers for T2DM diagnosis. Vastrad et al 11 utilized expression profiles of T2DM and normal control datasets along with PPI network and functional enrichment to identify plausible candidates for T2DM and their mechanistic pathways. The study also predicts potential miRNA and transcription factors involved in regulatory functions, identifying JUN, RELA, U2AF2, VCAM1, FN1, CDK1, ADRB2, TK1, ACTA2 andA2M as potential bio-markers for T2DM. In a similar study utilizing expression profiles of diabetic obese and non-diabetic obese samples, Vastrad et al 12 also identified key genes such as FLNA, MYH9, CLTC, ERBB2, DCTN1, TCF4, VIM, LRRK2, CAV1 and IFI16 as novel, probable biomark-ers for progression of obesity associated T2DM.
There is abundant literature on integrating differential gene expression profiles with network analysis and functional enrichment to describe molecular mechanisms of T2DM. However, there are numerous T2DM-associated genes identified through candidate gene studies, Genome Wide Association Studies (GWAS) and Exome/Genome Sequencing (WES/WGS). Studies aimed at leveraging these T2DM-associated genes to establish their mechanistic roles in disease development and progression would be insightful. Prioritization of potential candidates from established disease-associated genes using network based approaches is rapidly gaining popularity as it provides time and cost efficient alternative to biological validation of all the T2DM-associated genes identified thus far 13-15. In view of this, we employed integrated network analysis, gene set enrichment and pathway interrelation approaches to discern possible candidates from T2DM-associated genes.
Materials and Methods :
Compilation of gene set
T2DM associated genes were extracted from two publicly available databases: Type 2 Diabetes Know-ledge Portal by the Genetics of Type 2 Diabetes Consortium (GoT2D) 16,17 and T-HOD database 18. T2D knowledge portal lists predicted effector genes for T2DM using a heuristic scoring method based on genetic, regulatory and perturbation evidences derived from large genomic studies. Genes predicted to have "causal", "strong" and "moderate" link to T2DM risk were considered for present study. T-HOD is a literature based database that provides information on candidate genes for hypertension, obesity and diabetes. Additionally, DisGeNET 19 was also used to retrieve curated genes for T2DM. Genes from all three sources were collated and a list of non-redundant, unique genes was compiled.
Construction of protein-protein interaction network and hub genes identification
PPI network was constructed using STRING v11.0 (Search Tool for the Retrieval of Interacting Genes/ Proteins) (https://string-db.org/) 20. The compiled list of genes was used as input with Homo sapiens as the organism of interest. Experiments, databases and co-ex-pression were used as active interaction sources, with a median confidence score of >0.4. Rest of the parameters were set to default. The resultant protein interaction network was visualized in Cytoscape v3.8.0.0 21. Significant hub genes from the large network were identified using cytoHubba application 22. Top 10 genes evaluated employing five topological analysis methods namely, Degree, MCC, MNC, EPC and EcCentricity were selected for further analysis. The intersecting genes derived using these five algorithms encodes core proteins and may represent key candidate genes with important biological functions.
Gene set enrichment and pathway interrelation analysis
Functional annotation was performed using the ontologies and pathways function on Enrichr 23-25 to categorize the top scoring genes into biological processes, molecular functions, cellular components and pathways. Enrichment terms relevant to T2DM and associated phenotypes with a significant p-value of <0.05 were considered. ClueGO 26 was used on Cytoscape for pathway interrelation analysis using KEGG annotations. Right-sided hypergeometric test with Benjamini-Hochberg method for p-value correction was employ-ed.
Results :
Compilation of gene set
The T-HOD database yielded a total of 497 T2DM specific genes while 79 predicted effector genes were extracted from GoT2D knowledge portal. A total of 160 curated genes were retrieved from DisGeNET database. All three gene sets were combined and duplicate gene symbols were removed to obtain 615 unique, non-redundant genes used to build PPI network.
Construction of protein-protein interaction network and hub genes identification
PPI network constructed from 615 seed genes on STRING database yielded a complex network of 762 interactions among 605 nodes (Figure 1). The resultant edges were found to be more than the expected interact-tions indicating that the queried proteins are at least
partially biologically connected as a group. Importing the PPI network to Cytoscape and analyzing using de-gree, MCC, MNC, EPC and EcCentricity methods on cytoHubba resulted in identification of 23 top scoring hub genes (Figure 2A-E). IGF1R and EGFR were observed to be intersections of the five algorithms as shown in the Venn plot (Figure 3) and may serve as possible candidates for further studies.
Gene set enrichment and pathway interrelation analysis
Functional association of the top scoring hub genes with biological processes, molecular function, cellular components and pathways performed on Enrichr revealed significant enrichment in pathways for insulin resistance, type 2 diabetes, signaling cascades such as insulin receptor signaling pathway, PI3K signaling, IGFR signaling pathway, ERBB signaling pathway, MAPK signaling pathway and their regulatory mechanisms (Table 1). Additionally, the hub genes were also overrepresented in pathways related to vascular complications associated with diabetes such as diabetic cardiomyopathy, AGE-RAGE signaling pathway in diabetic complications, fluid shear stress and atherosclerosis, cellular response to lipid and atherosclerosis (Table 2). The interrelation between these pathways and 23 hub genes identified was explored using Cytoscape ClueGO and is depicted in figure 4.
Discussion :
The steady increase in prevalence of T2DM over the last few decades has led to diverse efforts towards delineating its molecular mechanism and pathophysiology. Numerous causal and susceptibility genes associated with T2DM have been identified by several genetic studies over the years. However, owing to the complex etiology of T2DM, unraveling the precise molecular pathways involved in disease development and progression has been challenging. In this view, complex diseases are progressively being studied using systemic network approaches combined with functional analysis to decipher the mechanism of action of genes involved 5,7.
In recent years, gene ontology and network based approaches have been employed extensively for T2DM biomarker identification using enormous data from linkage studies, GWAS and more recently-NGS based genome and transcriptome studies. PPI networks constructed using genes associated with T2DM and T2DM-related phenotypes are used to identify hub genes that possibly have a role to play in disease pathophysiology. In this study, we implemented integrated PPI network and hub gene analysis on 615 T2DM associated genes, along with gene set enrichment and path-way interrelation analysis to identify 23 genes namely, AKT1, ARRB2, CAV1, CTNNB1, DOK5, EGFR, ESR1, GSK3B, IGF1R, INSR, IRS1, IRS2, MAPK14, MAPK8, PIK3R1, PIK3R2, PRKAA1, PRKAA2, PRKCD, PTPN-11, SHC1, SIRT1 and SOCS that are plausible high-risk candidates for T2DM.
Of these, two tyrosine kinase receptor genes-EGFR and IGF1R were identified as common nodes by all five algorithms of cytoHubba and can be considered to be significant candidate genes in T2DM. Epidermal Growth Factor Receptor (EGFR) is a transmembrane glycoprotein that holds a key role as signaling molecule in the regulation of functions such as cellular growth, migration, proliferation and survival. Abnormal expression and signaling of EGFR is associated with diabetes, cardiovascular disease and cancer 27. There is growing body of research that suggests EGFR may play dual roles in the pancreas wherein it is integral for the regulation of pancreatic β cell mass and also mediate development of detrimental pathologies such as pancreatic fibrosis 28. Elevated EGFR signaling and increased receptor tyrosine kinase activity results in the dysfunction of diabetic micro-and macro-vas-culature 29,30. EGFR also mediates detrimental pathways involving Endoplasmic Reticulum (ER)-stress, Reactive Oxygen Species (ROS) and Renin-Angio-tensin-Aldosterone-System (RAAS) systems, leading to diabetic cardiac myopathy. On the other hand, EGFR is believed to be beneficial in the non-diabetic heart, with key roles in cardiac development and survival of adult myocardium 31. Insulin-like Growth Factor-1 Receptor (IGF1R) facilitates the action of Insulin-like Growth Factor-1 (IGF1) by activating the PI3K/ AKT and MAPK signaling pathways. IGF1R signaling is important in several physiological functions during cellular growth, differentiation and survival in normal and diseased conditions such as cancer and diabetes-induced cardiovascular dysfunction 32. IGF1 has been demonstrated by several independent studies to have a role in diabetic vascular diseases such as, atherosclerosis, angiogenesis, hypertension and restenosis 33. In diabetic conditions, Advanced Glycation End-products (AGEs) are formed as a result of prolonged hyperglycemia. Vascular proliferative changes that are induced due to hyperglycemia are reported in human monocytes as a consequence of AGE-induced IGF1 synthesis 34. Studies on diabetic APOE-null mice have de-monstrated that AGEs promote atherosclerotic lesion formation by enhancing proinflammatory pathways 35. Two distinct hallmarks of atherosclerosis–Vascular Smooth Muscle Cell (VSMC) differentiation and migration leading to atherosclerotic plaque formation, and destabilization and rupture of the plaque as a result of VSMC apoptosis are also intricately governed by differential expression of IGF1 and IGF1R 36-38.
Pathway interrelation analysis on ClueGO identified crosstalk between hub genes and molecular mechanisms which are generally known to underlie T2DM pathogenesis. Functional enrichment analyses on Enrichr identified several crucial processes and signaling pathways that are key factors for disease development. In particular, over-representation was seen in biological processes related to insulin receptor signaling, PI3K signaling, response to insulin stimulus, IGFR-signaling process and their regulation; molecular functions such as IGFR binding, serine/threonine kinase activity and protein kinase binding.
The past few decades have seen abundant efforts towards understanding the role of these signaling path-ways in T2DM. Circulating insulin, insulin and insulin-like growth factor receptors, and their corresponding signal transduction hold pivotal roles in glucose and energy metabolism. Under physiological conditions, insulin signaling cascade gets triggered by the binding of insulin to its receptors (IR), which undergo autophosphorylation and recruit IR substrates. Downstream signaling partners are then activated through kinases such as FOXO, AMPK, GSK3, ERK, mTOR, PI3K and AKT 39-42. Dysfunction of these pathways could lead to abnormal glucose homeostasis and subsequent insulin resistance, which is the primary cause of T2DM.
Of these, PI3K plays an important role in insulin action by activating the PI3K/AKT cascade. The PI3K/ AKT signaling pathway gets activated by postprandial insulin secretion, leading to increased utilization of glucose and reduced gluconeogenesis in muscle and liver. Improper functioning of PI3K/AKT insulin signaling pathway results in insulin resistance, leading to reduced functioning of β-cells 43. Enrichment analysis also revealed significant enrichment of genes in pathways such as diabetic cardiomyopathy, AGE-RAGE signaling pathway in diabetic complications, cellular response to lipid, fluid shear stress and atherosclerosis indicating possible involvement of these genes in development of diabetic cardiovascular complications. In our previous work 44, we have described in detail the mechanistic action of these processes leading to the development of coronary artery disease. Thus, as established by pathway interrelation analysis, a complex interplay of genes is involved in the functioning of these pathways. Therefore, any disruptive change in these genes and pathways may trigger a series of pathological outcomes.
Conclusion :
In the current study, we employed sequential network and pathway analysis to identify 23 plausible high risk candidate genes for T2DM and its involve-ment in functional processes leading to disease development. Two genes, EGFR and IGF1R can be considered significant candidates for further exploration and understanding with respect to T2DM. Thus, it can be derived that the onset of T2DM and its progression might be a consequence of disrupted downstream processes caused by any perturbations in the normal functioning of these identified genes. The study is aided by ontology based gene filtration and prioritization. However, these results serve as preliminary data for subsequent research on T2DM etiology. As in silico analyses alone cannot provide substantial evidence, these results need to be validated through appropriate wet-lab experiments.
Acknowledgement :
We are grateful to Department of Studies in Genetics and Genomics, University of Mysore for providing facility to conduct this work. We also thank members of the Genetics and Genomics lab, Department of Studies in Genetics and Genomics for their support and encouragement. We thank Indian Council of Medical Research (ICMR) for providing fellowship to Mrs. Tejaswini Prakash.
Conflict of Interest :
The authors declare no competing financial interests.

Figure 1. Protein interaction network constructed on STRING database using T2DM-associated genes.
|
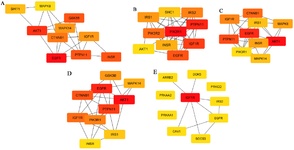
Figure 2. Top ranking gene clusters derived from cytoHubba. Algorithms used: A) Degree, B) MCC, C) MNC, D) EPC, E) EcCentricity.
|
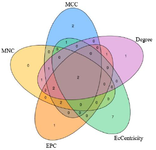
Figure 3. Venn plot depicting intersection of all five algorithms used on cytoHubba, with each algorithm represented by different colours: dark blue-MCC; purple-Degree; light blue-EcCentricity; orange-EPC; yellow-MNC. Number of genes found in common across algorithms is indicated. Two hub genes, EGFR and IGF1R, are found to be common across all the algorithms.
|
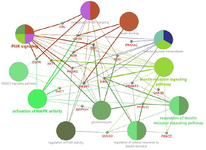
Figure 4. Pathway interrelation analysis of genes derived from top ranking gene networks. The enriched pathways are represented by larger nodes, and genes by smaller nodes. The corresponding edges indicate crosstalk between the enriched pathways and genes.
|
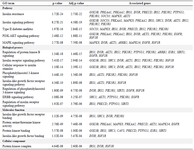
Table 1: Gene set enrichment and pathway analysis of 23 hub genes identified. Gene ontology categories: pathways, biological processes, molecular function, cellular component
|
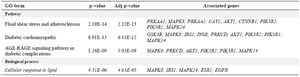
Table 2. Enriched pathways related to vascular complications
|
|