Designing a Multi-Epitope Antigen for Serodiagnosis of Strongyloides stercoralis Based on L3Nie.01 and IgG Immunoreactive Epitopes
-
Movahedpour, Ahmad
-
Department of Medical Biotechnology, School of Advanced Medical Sciences and Technologies, Shiraz University of Medical Sciences, Shiraz, Iran
-
Student Research Committee, Shiraz University of Medical Sciences, Shiraz, Iran
-
 Mostafavi-Pour, Zohreh
Department of Biochemistry, Shiraz University of Medical Sciences, zmostafavipour88@yahoo.co.uk
Mostafavi-Pour, Zohreh
Department of Biochemistry, Shiraz University of Medical Sciences, zmostafavipour88@yahoo.co.uk
-
Recombinant Protein Laboratory, Department of Biochemistry, School of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
-
Autophagy Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
-
Sarkari, Bahador
Department of Parasitology and Mycology, School of Medi-cine, Shiraz University of Medical Sciences, sarkarib@sums.ac.ir
-
Department of Parasitology and Mycology, School of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
-
Basic Sciences in Infectious Diseases Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
-
Taheri-Anganeh, Mortaza
-
Cellular and Molecular Research Center, Cellular and Molecular Medicine Institute, Urmia University of Medical Sciences, Urmia, Iran
-
Nezafat , Navid
-
Pharmaceutical Sciences Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
-
Savardashtaki , Amir
-
Department of Medical Biotechnology, School of Advanced Medical Sciences and Technologies, Shiraz University of Medical Sciences , Shiraz, Iran
-
Infertility Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
-
Ghasemi, Younes
-
Pharmaceutical Sciences Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
Abstract: Background: Serological diagnosis of Strongyloides stercoralis (S. stercoralis) is fre-quently challenging because of cross-reactivity with other parasitic nematodes. There-fore, it is necessary to introduce novel serological tests with high performance to properly diagnose this neglected parasitic infection. The purpose of the current study was to design a multi-epitope construct for the diagnosis of S. stercoralis.
Methods: For the purpose of this study, first, highly antigenic segments and potential immunodominant epitopes of S. stercoralis were identified from two antigenic proteins, and then all of the selected parts were linked by an appropriate linker. Next, the physico-chemical features of the designed construct were analyzed. Then, tertiary structures of the construct were built and evaluated to find out the best one. Lastly, the amino acid sequence was reverse-translated and optimized for over-expression in Escherchia coli (E. coli).
Results: The bioinformatic evaluation indicated that the designed protein construct could be hydrophilic, thermostable, and acidic and the estimated half-life was more than 10 hr in E. coli.
Conclusion: According to the results of the study, the designed construct could be used as an efficient antigen in the ELISA system for serological diagnosis of human strong-yloidiasis.
Introduction :
Strongyloides stercoralis (S. stercoralis) is a pathogenic neglected parasitic roundworm that causes human strongyloidiasis. This soil-transmitted helminth has spread throughout the world, especially in tropical and subtropical regions 1. Currently, S. stercoralis infects about 370 million people in the world 1,2. The mortality rate in patients with hospitalization is 16.7% and in immunocompromised patients is as high as 60-85% 3. The
S. stercoralis life cycle is complex and includes direct, indirect, and auto-infective ones 4. Regarding the prop-erty of the auto-infective cycle, the parasite remains undiagnosed for years and causes a chronic disease that resists treatment 5. Nearly, half of the patients with strongyloidiasis are asymptomatic, which results in the development of a chronic disease due to the unique life cycle of the parasite 2.
Hyperinfection and distribution can be seen in high-risk groups, including patients undergoing glucocorticoid therapy, patients with hematologic malignancy, co-infected patients with human T Lymphotropic Virus type I (HTLV-1) and HIV, and those with chronic alcohol abuse 6. Diagnostic methods for strongyloidiasis can fall into two main categories of serological and non-serological approaches, and often a mixture of both methods is required for accurate and proper diagnosis. Non-serological systems are based on intestinal parasite recognition, molecular methods, and antigen detection, while the serological approaches are based on the finding of anti- S. stercoralis antibodies 5,7.
Due to the parasitic load and low severity of infection, it is difficult to definitively diagnose strongyloidiasis by stool examination, so the sensitivity of this method is very low. Although molecular methods are more sensitive, there are differences in the accuracy of PCR reports 8,9. A few serological tests are presently commercially accessible to diagnose strongyloidiasis, but possible cross-reactions with other parasites have been considered an obstacle in this case. Therefore, the application of recombinant and synthetic antigens has been suggested as suitable options for diagnosing this disease 10.
The advent of in silico methods has provided a platform for predicting the molecular structures and investigating the receptors and ligands interactions 11,12. Today, computer science and information technology are utilized to analyze biological information such as DNA decoding and predicting the structure and function of proteins, and accordingly, a new field called science bioinformatics is being developed 11,13-16. Studies show that bioinformatics can help biological and medical studies by saving time, decreasing the number of experiments, and designing novel biological molecules with desirable features 17-21. Computational immunology is a newly introduced field, using computer science in immunology. Today, computational immunology can be used in various fields, including identification of potential antigens, estimation of B and T cell epitopes, and designing various forms of vaccine. The use of computational tools can decrease the time and cost in experimental procedures 22. Considering the advantages mentioned above, the purpose of the present study was computational designing of a multi-epitope antigen originated from two main antigens of S. stercoralis to diagnose human strongyloidiasis.
Materials and Methods :
Study design
In the current study, firstly, the proteins with antigenic properties were selected. Then, using the bioinformatics softwares, the epitopic areas of B cells were selected. The designated epitopes were linked using appropriate amino acid linkers and the 3D model of the ultimate structure was constructed and validated by in silico tools. Lastly, for the expression of the designed structure in the Escherichia coli (E. coli) host, the amino acid sequence was reverse-translated to the DNA and optimized.
Estimating antigenicity
EMBOSS server v 6.6.0.0 (available at http://www.
bioinformatics.nl/cgi-bin/emboss/antigenic) was employed to predict the antigenicity features of the proteins 23. EMBOSS is an algorithm that contains several programs for various analyses, for instance, alignment of sequences, searching databases, recognition of motifs in proteins, prediction of patterns in nucleotide sequences, and analysis of codon usage 24,25. Also, it identifies antigenic parts of proteins according to physico-chemical properties, amino acid composition, and epitope redundancy.
In the present work, default parameters (minimum length of the antigenic region=6, output report format=EMBOSS motif) were set up to define the antigenic segments of the selected proteins 23.
Linear B cell epitopes
Epitopes are regions of the antigen that are detected by the paratope region of the immunoglobulin. The linear epitope is a continuous amino acid sequence. The conformational epitope consists of amino acids that may not be in the same sequence but reside near each other in a 3D structure. A critical step in designing a multi-epitope protein for vaccine production or building a serological test is the prediction of B cell epitopes. Thus, bioinformatics methods are desirable for prediction of linear B cell epitopes in proteins. In addition, bioinformatics approaches can provide cost and time-saving methods for predicting probable B cell epitopes in a target protein 26.
The linear antigenic B cell epitopes were identified via BCPred v2.0 (available at http://ailab.cs.iastate.edu/ bcpreds/predict.html), an innovative way for linear B cell epitopes estimation by the subsequence kernel. The BCPred resource was applied for identification of the B cell epitopes with a size of 20 amino acids, set to 75% specificity. Furthermore, the server uses an innovative approach of a subsequence kernel with 74.57% accuracy. Then, the chosen variants of each antigen are submitted in plain form, and approximately 4 to 7 segments are predicted as the final epitopes 27.
The linear B cell epitopes are also estimated by ABCPred tools of immune epitope database (available at http://crdd.osdd.net/raghava/abcpred/) with default settings. The best predictions by ABCPred occur when 16-mer peptide (ABCP16) is set up 28. ABCPred server can define B cell epitopes in the proteins through artificial neural networks. Furthermore, ABCPred is the primary online software established according to the recurrent neural network by fixed-length patterns with 65.93% accuracy 27.
Designing the construct
The B cells epitopes that were selected by the aforementioned servers were fused via appropriate amino acid linkers. Finally, an amino acid construct was built based on a multi-epitope approach.
Assessment of the physico-chemical features
Stability and efficiency of a protein structure in a biological system are related to their physico-chemical properties. Numerous physico-chemical features including amino acid sequence, extinction coefficient, instability index, grand average of hydropathicity index (GRAVY), aliphatic index, isoelectric point (pI), and molecular weight can help predicting the stability, activity, and overall properties of the protein 29. There are various online softwares that can determine the physico-chemical features of a given protein.
ProtParam tool (available at http://web.expasy.org/ Protparam/) is an online software at ExPASy server that was utilized in our study to analyze different physico-chemical properties of our designed construct, based on its amino acid sequence 30.
In silico cloning
The OPTIMIZER (available at http://genomes.urv. es/OPTIMIZER) was utilized for reverse translation of amino acid to DNA and codons to prepare the optimized DNA sequence of the construct for cloning and expression in E. coli host. Moreover, critical parameters of the final optimized nucleotide sequence, including GC content, Codon Adaptation Index (CAI), and Codon Frequency Distribution (CFD) were analyzed by Gen Script rare codon analysis (available at https://www.genscript. com/tools/rare-codon-analysis) 13.
Building 3D model for the construct
The 3D structures of proteins can help to predict the protein functions and provide useful information for designing the regulator molecules that can regulate the functions of proteins. For 3D structure of our construct, the GalaxyTBM (Template-based modeling, available at http://galaxy.seoklab.org/) was applied 31.
TBM, also termed homology modeling or comparative modeling, usually consists of the following steps 32,33:
1- adopting the homologous proteins with experimentally defined structures as templates; 2- alignment of the target sequences and templates; 3- building the 3D model, based on the alignment; and 4- refinement of the models. Current methods regularly treat each step distinctly, and the full TBM process can then be set up by merging methods for each of the above steps 31.
Primarily, templates are chosen by rescoring HH-search results which assign more weights for the score of secondary structure. Among the re-ranked top 20 homologs, various templates are chosen via eliminating structural deviations according to mutual TM scores for kernel alignment 34,35.
PROMALS3D is utilized for multiple sequence alignment of kernel segments removing unaligned termini 29. Alignments of terminal sequences are attached afterward. Primary model structures are then constructed based on the templates and the alignment via CSA (conformational space annealing) for total optimization of the regions, originated from templates 36,37.
Tertiary structure validation
A significant step for discovering the biological procedures at a molecular level is the availability of a structural model of the protein. After processing, preparing, and reviewing the 3D model using online softwares such as ProSA, RAMPAGE, and ERRAT, the constructed model's quality and reliability were assessed 38,39. A critical online software, used to examine the 3D models of protein for possible errors, is ProSA 40. This tool is utilized in the diagnosis of empirically identified structures 34,35, theoretical models 36, and protein engineering 41. ProSa z-score can determine the 3D models' quality (http://prosa.services.came.sbg.ac.at/prosa.php) 42. The total quality score assessed by ProSA for a definite input structure is shown in a plot that indicates the scores of all empirically defined protein chains, available in the Protein Data Bank (PDB) 43. This property connects the score of a specific model to the scores calculated from all defined structures that are saved in PDB. Hard segments of a 3D structure are known by a plot of local quality scores and the same scores are mapped on a display of the tertiary structure, using color codes. ProSA can help for validating 3D protein structures before submitting to PDB 44.
Moreover, the quality of generated models was evaluated with PROCHECK by Ramachandran plot (available at http://mordred.bioc.cam.ac.uk/rapper/rampage. php) 45,46. Ramachandran Plot (RP) indicates each pair of (φ, ψ) angles in 3D structure of a specified protein in a simple logical way 13. Ramachandran plot has long been employed in every step of crystallography and protein modelling to identify the main chain torsion angles (φ,W) 46.
Also, the package calculates the Ramachandran angles at the central residue in the stretch of three amino acids which have definite types of flanking residues. The package shows the Ramachandran plots with a thorough analysis of output. This package is accommodated with all the 3D structures accessible in the Protein Data Bank (PDB) 47.
The ERRAT (available at http://services.mbi.ucla. edu/ERRAT/) is a tertiary structure validation algorithm for analyzing the procedure of crystallographic model construction and refining preserved by UC Global Health Institute, University of California, USA 48. The ERRAT explains the statistics of non-bonded interaction between different atoms, and a score of 50 is commonly appropriate for the tertiary structure assessment 49. In the current research, six types of noncovalently bonded atom-atom interactions (CC, CN, CO, NN, NO, and OO) in 3D models were measured 48.
Molecular dynamics simulations
Molecular Dynamics (MD) simulations were carried out by GROMACS 2019.6 package 50, using the Amber99sb force field 51. At first, multiepitope antigen was placed in a triclinic simulation box (6.20700 nm× 18.54500 nm×11.35100 nm) and filled with water molecules using TIP3P model 52.
To confirm the total charge neutrality of the simulated systems, sufficient amounts of counter-ions were added instead of water molecules keeping physiological salt concentration at 0.15 M. Periodic boundary condions were applied in three spatial dimensions. An energy minimization process was carried out using the steepest descent method to remove large forces and relax the systems 53.
The first phase of equilibration involved 500 ps stimulation under the canonical ensemble (NVT) followed by a 500 ps NPT equilibration. The temperature was maintained at 277 °K (refrigerator temperature) and room temperature (298 °K) and the pressure was maintained at 1.0 bar with the Berendsen thermostat and barostat 54. In total, after two phases of equilibrations, 150 ns molecular dynamics simulations were done to record trajectories using the leap-frog algorithm and solve the equations of motion with a time step of 0.002 ps. The Particle Mesh Ewald (PME) method, Lennard-Jones potential, and LINCS algorithm were used for computation of long range electrostatics, van der Waals interactions, and covalent bond constraints, respectively 55-57.
Results :
Sequence retrieval
The amino acid sequences of L3Nie.01 and IgG immunoreactive antigens in FASTA format were obtained from the NCBI database (Table 1).
Immunoinformatic analyses
The EMBOSS server showed the most antigenic sites, based on the score of each residue (Table 2).
Defining linear B cell epitopes
According to BCPRed and ABCPred, the linear epitopes of each antigen were predicted. Thus, the epitope sequences for each of the antigens, along with their primary and terminal amino acids, the number of amino acids, and the score for each sequence are shown in table 3. Then, using Microsoft Excel software, the epitopes mentioned above were chosen. The final selected segments were again compared with the segments in the CLC Genomics program for the two antigenic proteins; both the conserved regions and the best immunogenic proteins were chosen. In the last step, the multi-epitope protein was designed, using the appropriate linkers between selected regions. The finally selected regions of L3Nie.01 and IgG immunoreactive proteins are presented in table 4.
Multi-epitope construct
The selected B cell epitope regions of L3Nie.01 and IgG immunoreactive proteins, shown in table 4, were fused via the EAAAK linkers, and the multi-epitope construct was designed.
Evaluation of the physico-chemical features
The physico-chemical features of the designed structure were calculated by ProtParam. The Molecular Weight (MW) and pI value of the designed protein were 26.8 kDa and 5.05. The pI value showed the acidic nature of the protein. Also, the construct is composed of 237 amino acids. The predictable half-life of the designed construct was 1 hr (mammalian reticulocytes in vitro), 30 min (yeast in vivo), and >10 hr (E. coli in vivo). The Instability Index (II) was found to be 28.04, which showed that the construct could be considered as a stable protein. Aliphatic index and GRAVY value were calculated as 54.47 and −1.378, which indicated the hydrophilic nature of the construct and showed that the protein can interact with water molecules.
Optimization of codons
Reverse-translation and codon optimization were done by the online software for over-expression in E. coli. The final optimized sequence was analyzed via the GenScript. The results indicated that the CAI of the sequence was 0.94. The CAI of 1.0 is ideal and CAI >0.8 could be considered optimal for expression in the bacterial host (Figure 1A). The GC content of the gene was 53.65% (Figure 1B). A GC content in range of 30% to 70% is a suitable value. All peaks outside this range would have an adverse effect on transcriptional and translational efficiency. Codon Frequency Distribution (CFD) of 91-100, 81-90, 71-80, and 31-40 in the gene was 81, 5, 10, and 1%, respectively (Figure 1C).
3D modeling, refinement, and validation of the tertiary structure
The GalaxyTBM created five 3D structures. Then, ERRAT, ProSA, and Ramachandran plot servers were utilized for analyzing the quality of tertiary models and choosing the best one (Figures 2-4). The probable errors of the five models were determined by ProSA with a z-score, which indicates similarity of the target 3D structure and experimentally defined structures (Table 5). ProSA computes the total quality score (z-score) of the protein and compares it with experimentally defined structures of proteins in the same size according to PDB results. Positive values show the problem in the created 3D models and the highest negative z-score indicates the highest quality of tertiary structure of the designed construct. The results of the Ramachandran plot and ERRAT analysis for five models are illustrated in table 3. After comparing tertiary models, based on the results of the aforementioned servers, model 3 was considered as the most appropriate model. The ERRAT quality and ProSA z-score were 95.755 and -3.32 for model 3. Furthermore, the Ramachandran plot indicated that 92.7, 6.8, and 0.5% of the amino acid residues were situated in the favored region, allowed region, and outlier region, respectively.
The results of molecular dynamics simulations
MD simulation study assists us to understand structure and dynamics of multiepitope antigen with extreme details. For this purpose, GROMACS analysis tools were used to analyze the trajectories in terms of Root Mean Square Deviation (RMSD), Root Mean Square
Fluctuation (RMSF), Radius of gyration (Rg), and secondary structure.
Root mean square deviation (RMSD)
To confirm the stability of the simulations, the RMSDs of multi-epitope antigen at room and refrigerator temperatures were analyzed during 150 ns simulation time. As seen in figure 5, RMSD plot of multi-epitope at 298 K increased gradually and reached equilibrium around 75 ns and after that remained stable with low RMSD fluctuations. High value of RMSD fluctuations were seen at 277 °K but finally it reached equilibrium around 120 ns and thereafter remained stable with low RMSD fluctuations.
Radius of gyration (Rg)
The compactness of multi-epitope antigen structure during the MD simulation time was assessed by analysis of the Rg. As seen in figure 6, the radius of gyration of multi-epitope antigen at 298 °K decreased gradually which represents the compact structure of multi-epitope antigen. Finally, it reached to equilibrium around 75 ns and after that remained stable with low fluctuations. High value of Rg fluctuations was seen at 277 °K showing the somewhat unfolded multi-epitope antigen at 277 °K but it reached equilibrium around 120 ns and thereafter its fluctuation was unnoticeable.
Root mean square fluctuation (RMSF)
The analysis of RMSFs of multi-epitope antigen can be used as a reference to estimate flexibility of the residues (Figure 7). The results of the RMSF analysis of multi-epitope antigen showed that the highly fluctuating areas appeared in the random coil structures. Moreover, some residues have a higher RMSF value at 277 °K than 298 °K, showing the partially unfolded multi-epitope antigen at 277 °K.
Secondary structure
The secondary structure of the multi-epitope antigen was calculated with the DSSP code of GROMACS. Figure 8 shows the α-helix, β-sheet, and other secondary structures of the multi-epitope antigen in two simulations. As seen in this figure, at two temperatures, the main secondary structures were rather stable and major conformational change of multi-epitope antigen was a simple motion during the 150 ns simulation time.
Discussion :
Strongyloidiasis is usually difficult to diagnose as the larval output is irregular and the parasite load is low. Serological methods, based on the detection of anti-S. stercoralis antibodies, are believed to be more applicable.
However, the cross-reactivity with other parasites antigens decreases the performance of serological tests for the diagnosis of strongyloidiasis 58. Today, ELISA and related methods are commonly used serological tests due to low costs, easy use, less variable results, and ease of automation 59. Using recombinant DNA technology, all protein antigens could be easily produced and applied for designing an ELISA kit 60-62. Diagnostic ELISA kits based on selected epitopes of B cells are novel and efficient approaches for serological diagnosis of infectious diseases 63,64.
Nowadays, bioinformatics tools and approaches help to design protein molecules with desirable and appropriate features 65-68. In silico methods have revolutionized the field of medical biotechnology by presenting a platform via predicting molecular structures and the molecular interactions between the designed therapeutics and their potential target receptors 69. Thus, online bioinformatics tools were used to design a multi-epitope protein for the diagnosis of strongyloidiasis, based on B cell linear epitopes of L3Nie.01 and IgG immunoreactive antigens.
Bisoffi et al showed that the L3Nie.01 antigen has no cross-reactivity with other parasite antigens 70. Furthermore, it has been shown that the diagnostic sensitivity of recombinant L3Nie.01 antigen is in the range of 72.3 to 78.9%, and its specificity ranges from 85.1 to 93.6% 58. Therefore, L3Nie.01-ELISA has been suggested as a serological method for assessing and determining treatment outcomes in endemic areas and has a significant role in monitoring public health interventions against strongyloidiasis 71.
Linker design is a very important and fundamental principle in the construction of de novo designed multi-domain proteins. Controlling the distance and orientation of domains in order to maximize the function of the desired protein is a difficult task in protein engineering. In 2001, Arai et al engineered and designed a suitable linker that aimed to provide logical consistency, flexibility, and spacing between the domains of a multifunctional fusion protein. It is shown that EAAAK helix binders can very effectively separate fusion protein domains, and that distances between domains can be controlled by changing EAAAK binding motifs. Therefore, the high potential of helical binders was indicated to be useful in protein engineering and design of multifunctional fusion proteins 72. In the present study, two servers were utilized for enhancing the prediction accuracy of linear epitopes with 20-and 16-mer B cell epitopes. Then, Microsoft Excel software was used to detect the most common and overlapping regions of each chosen protein. The selected regions of linear B cell epitopes were fused via EAAAK linker and a novel multi-epitope construct was built. To fix the distance between functional domains and intensify the expression of the final product in the bacterial host, EAAAK helical linker with the support of Glu-Lys salt bridge created a coherent structure 73,74. The absence of the linkers between the epitopes leads to the production of connected epitopes (neo-epitopes) and disturbance of protein function 75. Farhani et al utilized EAAAK as a rigid linker for binding HTL epitopes in their construct and designed a stable multi-epitope vaccine against pathogenic Shigella spp 76. Furthermore, Vakili et al used EAAAK peptide linker for binding multi-epitope construct of Leishmania infantum antigens to peptide adjuvant in N- and C- terminals which results in making a stable structure 74.
The DNA sequence of the construct was optimized based on codon usage of E. coli host. Analysis of optimization parameters including CAI, CFD, and GC contents by GenScript server showed that the optimized nucleotide sequence was suitable for overexpression in E. coli host.
The physico-chemical features of the designed construct were also predicted via the bioinformatics tool. The construct was hydrophilic based on the GRAVY score -1.378, which demonstrates its easy interaction with water molecules. The pI value 5.05 showed the acidic feature of the designed protein. Besides, the high aliphatic index 54.47 showed that the designed protein could be more thermostable, and also, the value of the instability index 28.04 revealed that the designed construct could be stable. The 3D model building and validation can open a horizon to identify the structure of the proteins. For this reason, five 3D models of the construct were built by GalaxyTBM. Moreover, ProsA and ERRAT were utilized to evaluate and select the best model.
Conclusion :
In the present study, immunodominant linear B cell epitopes from two antigens of S. stercoralis, were predicted and linked together via proper linkers. The study of physico-chemical structures of designed protein construct showed that the structure is thermostable, hydrophilic, and acidic with a half-life of more than 10 hr in E. coli. Finally, it was found that the designed chimeric protein can express greatly in E. coli. The designed protein can be used in the construction of a high-performance serological test for the diagnosis of human strongyloidiasis.
Acknowledgement :
This study was financially supported by the office of vice-chancellor for research of Shiraz University of Medical Sciences (Grant No. 18780). The results described in this paper were part of PhD thesis of Ahmad Movahedpour.
Conflict of Interest :
The authors declare that they have no conflict of interest.

Figure 1. Analysis of three important features of the designed sequence for high-level protein expression in E. coli host. A. Codon Adaptation Index (CAI), B. GC content, C. Codon with the Frequency Distribution (CFD).
|

Figure 2. The ERRAT analysis results of models 1, 2, 3, 4, 5.
|
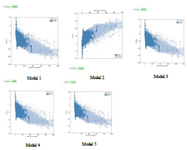
Figure 3. The ProSA scores of models 1, 2, 3, 4, 5.
|

Figure 4. The Ramachandran plot analysis of five models.
|
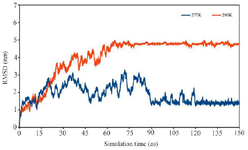
Figure 5. RMSD of multi-epitope antigen at 277 K and 298 K during total simulation time.
|
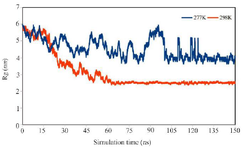
Figure 6. Time dependence changes of Rg of multi-epitope antigen during the simulation at 277 °K and 298 °K.
|
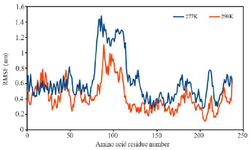
Figure 7. RMSF of residues of multi-epitope antigen from time-averaged positions during the last 30 ns at 277 °K and 298 °K.
|
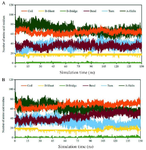
Figure 8. Variation of the secondary structure versus time for the multi-epitope antigen at a) 277 °K and b) 298 F.
|
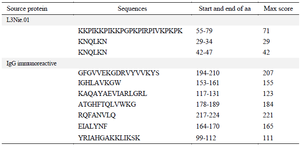
Table 1. Prediction of antigenic regions of L3Nie.01 and IgG immunoreactive proteins via EMBOSS server
|
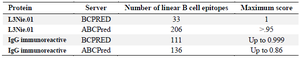
Table 2. Linear B cells analysis of L3Nie.01 and IgG immunoreactive by BCPRED and ABCPred
|

Table 3. Selected B cell epitopes regions of L3Nie.01 and IgG immunoreactive proteins
|
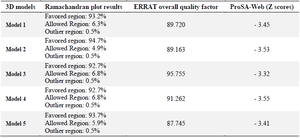
Table 4. The evaluation of five 3D structures via Ramachandran, ERRAT, and ProSA-Web servers
|
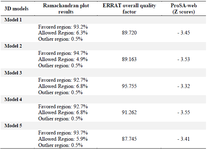
Table 5. The evaluation of five 3D structures via Ramachandran, ERRAT, and ProSA-Web servers
|
|