A Nonlinear Pattern Recognition of Pandemic H1N1 Using a State Space Based Methods
-
 S.Mabrouk, Mai
Mai S. Mabrouk, Ph.D., Biomedical Engineering Department, Misr University for Science and Technology (MUST), Egypt, E-mail: msm_eng@k-space.org
S.Mabrouk, Mai
Mai S. Mabrouk, Ph.D., Biomedical Engineering Department, Misr University for Science and Technology (MUST), Egypt, E-mail: msm_eng@k-space.org
-
Biomedical Engineering Department, Misr University for Science and Technology (MUST), -, Egypt
Abstract: Genomic Signal Processing is a relatively new field in bioinformatics, in which signal processing algorithms and methods are used to study functional structures in the DNA. An appropriate mapping of the DNA sequence into one or more numerical sequences enables the use of many digital signal processing tools in the analysis of different genomic sequences. Also, a novel Influenza A (H1N1) virus of swine origin emerged in the spring of 2009 and spread very rapidly among people. The severity of the disease and the number of deaths caused by a pandemic virus varies greatly and can change over time. Throughout this work, Pandemic H1N1 genomic sequences were characterized according to nonlinear dynamical features such as moment invariants and largest Lyapunov exponents and then compared to those features that extracted from classical H1N1 genomic sequences. The proposed methods were applied to a number of sequences encoded into a time series using a coding measure scheme employing Electron-Ion Interaction Pseudopotential (EIIP). The aim of this work is to extract genomic features that can distinguish the new swine flu from the classical H1N1 existed before using sequences from segment 8 of the influenza genome that consists of 8 RNA segments which encodes two important proteins for immune system attack (NS1 and NS2). According to the obtained results it is evident that variability is present based on a significance test in both groups; pandemic and classical H1N1 sequences.
Introduction :
The variations of pandemic H1N1 influenza virus are caused as a result of different mutations occurring during viral replication (1). The polymerase of this RNA virus lacks proof reading activity (2); this gives rise to considerable viral variability culminating in 3 different types A, B and C, in addition to many subtypes based on variations in the hemagglutinin (HA) and the neuraminidase (NA) surface proteins (3). The influenza genome consists of 8 RNA segments and encodes for 10 polypeptides; the internal structural proteins, nucleocapsid protein (NP), the two matrix protein (M) are used for the classification of the influenza virus into A, B and C. The surface proteins neuraminidase (NA) and hemagglutinin (HA) have been studied extensively and the antigenic variations in the these surface glycoproteins are used to subtype Influenza A. Additionally, three of the influenza polypeptides are associated with RNA polymerase activity (PA, PB1, PB2), and the RNA binding non-structural protein (NS) that contribute to viral pathogenicity and play a central role in the prevention of interferon mediated antiviral response. The Influenza A Virus (IAV) undergoes major and minor genetic variations, the yearly antigenic drift resulting in as minor as a single amino acid mismatch. Major variations known as antigenic shifts are the cause of serious outbreaks and pandemics as the 1918, 1957, and 1968 worldwide outbreaks (4). Changes in the genetic and antigenic composition result in challenges in the development of influenza vaccines and antiviral medications (5).
In the last two decades, there has been an increasing interest in applying techniques from the domains of nonlinear analysis and chaos theory in different fields of research. In this work, the chaos theory was applied to both pandemic H1N1 and classical H1N1genomic sequences in order to discriminate between them according to their non linear dynamical features as moment invariants, and Largest Lyapunov Exponents (LLE).
Materials and Methods :
The conversion of the DNA sequences into digital signals offers the possibility of applying signal processing methods to the analysis of genomic data (6,7). The genomic signal processing applications in bioinformatics provides an efficient tool used to extract features of DNA sequences maintained over the whole genomes (8). In this work, the EIIP sequence indicators were used, the energy of delocalized electrons in amino acids and nucleotides has been calculated as the Electron-Ion Interaction Pseudopotential (EIIP). The EIIP values of amino acids were used to substitute for the corresponding amino acids in protein sequences, whose power spectrum is taken to extract the information contents (9). To study the dynamics of the proposed system, the state space trajectory was first reconstructed. Phase space reconstruction is the fundamental for analyzing nonlinear signals, by which a time series can be embedded to n-dimensional space.
Briefly the basic steps of the reconstruction of the phase space were demonstrated. First, different sequences of the pandemic H1N1 and classical H1N1 which existed before were encoded into a time series signal using EIIP sequence indicators. A good choice for a delay time was yielded by using the first minimum of the auto mutual information function. The first minimum of the auto mutual information could be found at four. The minimal embedding dimension for the pandemic H1N1 and classical H1N1 time series signals were calculated using Cao's method with a delay time of four, a maximal dimension of eight, three nearest neighbors and reference point depending on the length of each signal. There was a kink produced by Cao's method at 3. This kink represents the time delay reconstruction of pandemic and classical H1N1 time series signals with embedding dimension of 3 and delay of 4. Finally, the phase space trajectory was obtained for both time series signals of the two types of H1N1 genomic sequences (pandemic and classical). The step following obtaining the phase trajectory is the step of feature extraction (10).
Feature extraction
TSTOOL software package is used to estimate the extracted nonlinear dynamical features; it is a software package for signal processing with emphasis on nonlinear time-series analysis (11).
Moment invariants
Features obtained by moment invariants are simple calculated features that do not change under translation, scaling or rotation (12). These invariants are constructed using the generalized fundamental theorem of moment invariants (GFTMI), which was formulated as in (13). The n-dimensional moments of order p of a function of intensity ρ (x1, ..., xn) = ρ (x) are defined in terms of Rieman integral as:
Where pi + ... pn = p, 0 < p < ∞. It is assumed that ρ (x) is piecewise continuous and therefore bounded function, and it can have nonzero values only in a finite part of the Rn; then the moments of all orders exist.
The central moments:
Where
The seven features of moment invariants:
Largest lyapunov exponent (LLE)
In this work, a set of genomic sequences from segment 8 of the influenza genome of both pandemic and classical H1N1 was downloaded from the NCBI. The length of these sequences was chosen to be 800-1000 bp. These sequences are first encoded using EIIP sequence indicators. Then, the phase space trajectory was reconstructed for each time series of both of them. The TSTOOL larglyap algorithm was used to estimate the Largest Lyapunov Exponent (LLE). This algorithm is similar to Wolf’s algorithm and provides an efficient estimation of the Largest Lyapunov Exponent through the calculation of the rate of increase of the prediction error versus the prediction time (14).
Result :
Results of moment invariants
Features based on moment invariants were computed after the construction of phase space of both pandemic and classic H1N1 EIIP encoded sequences. The seven features are arranged as (φ1, φ2, φ3, φ4, φ5, φ7, and φ8). A significance test (t-test) was performed on the proposed features to assess the use of such parameters for discriminating between them. The result of the t-test is presented and the p value is calculated for all seven features; they are all less than 0.05 as shown in table 1. Figure 1 shows the result of comparing the average features extracted based on moment invariants for pandemic and classical H1N1. There is a significant difference between the two types of H1N1 as shown in the figure. Also, small vertical bars represent a standard deviation across features.
Results of largest lyapunov exponent (LLE)
The LLE estimates of a set of pandemic and classical H1N1 genomic sequences were calculated using TSTOOL largelyap algorithm as shown in table 2. It is an algorithm very similar to the Wolf algorithm; it computes the average exponential growth of the distance of neighboring orbits via the prediction error. The increase of the prediction error versus the prediction time allows an estimation of the Largest Lyapunov Exponent. A significance t-test was applied to assess the use of LLE estimates in the discrimination between pandemic and classical H1N1.
Significance test
The accuracy of a test was evaluated to discriminate between pandemic H1N1 and classical H1N1 by moment invariants and Largest Lyapunov Exponent dynamical system features). These features were divided into three feature vectors as follows:
V1= {Ф1, Ф2, Ф3, Ф4, Ф5, Ф7, Ф8}
V2= {LLE}
V3= {Ф1, Ф2, Ф3, Ф4, Ф5, Ф7, Ф8, LEE}
The feature vectors were fed into the classification process using K-means clustering classifier. Results of applying the significance test are shown in table 3.
Discussion :
The proposed techniques were implemented and applied to a number EIIP encoded sequences of pandemic and classical H1N1 from segment 8 of the influenza genome to identify their genomic signatures as continuous detection of these signatures is important in the analysis of the adaptation process from nonhumans to humans.
Conclusion :
The analysis of different genomic mutations of the pandemic H1N1 genomic sequences is very important to study the possibility of virus adaptation from non- humans to humans.
Acknowledgement :
The author really expresses a deepest thanks to Dr. Yasser Kadah at the Department of Systems and Biomedical Engineering, Cairo University for his valuable discussions and continuous support; he is actively engaged in bioinformatics, including sequence analysis data mining, genomic signal analysis and software development. Also, the author presents many thanks to Dr. Mahmoud El-Hefnawi at National Research Center (NRC) for his support and help.
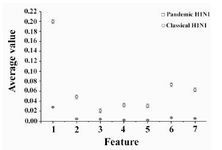
Figure 1. Features extracted based on moment invariants for pandemic and classical H1N1, the small vertical bars represent standard deviations across features
|
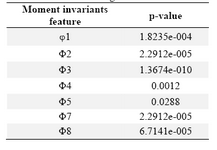
Table 1. P-value of t-test on a set of pandemic and classical H1N1 EIIP encoded sequences for feature extracted using moment invariants
|
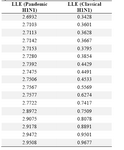
Table 2. Largest Lyapunov Exponent estimates of pandemic and classical H1N1 encoded sequences
|
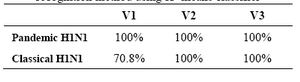
Table 3. Accuracy of the proposed nonlinear pattern recognition method using K- means classifier
|
|