Extraction, Purification and Characterization of Lipopolysaccharide from Escherichia coli and Salmonella typhi
-
Rezania, Simin
-
Department of Microbiology, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran
-
Amirmozaffari, Noor
-
Department of Microbiology, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran
-
Tabarraei, Bahman
-
Department of Bacterial Vaccine and Antigen Production, Pasteur Institute of Iran, Tehran, Iran
-
Jeddi-Tehrani, Mahmood
-
Monoclonal Antibody Research Center, Avicenna Research Institute, ACECR, Tehran, Iran
-
Reproductive Biotechnology Research Center, Avicenna Research Institute, ACECR, Tehran, Iran
-
Zarei, Omid
-
Monoclonal Antibody Research Center, Avicenna Research Institute, ACECR, Tehran, Iran
-
Alizadeh, Reza
-
Nanobiotechnology Research Center, Avicenna Research Institute, ACECR, Tehran, Iran
-
Masjedian, Faramarz
-
Department of Microbiology, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran
-
 Zarnani, Amir-Hassan
Amir Hassan Zarnani, D.M.T., Ph.D., Nanobiotechnology Research Center, Avicenna Research Institute, ACECR, Tehran, Iran, Tel: +98 21 22432020 Fax: +98 21 22432021 E-mail: zarnani@avicenna.ac.ir
Zarnani, Amir-Hassan
Amir Hassan Zarnani, D.M.T., Ph.D., Nanobiotechnology Research Center, Avicenna Research Institute, ACECR, Tehran, Iran, Tel: +98 21 22432020 Fax: +98 21 22432021 E-mail: zarnani@avicenna.ac.ir
-
Nanobiotechnology Research Center, Avicenna Research Institute, ACECR, Tehran, Iran
-
Immunology Research Center, Tehran University of Medical Sciences, Tehran, Iran
Abstract: Lipopolysaccharide (LPS) is an important structural component of the outer cell membrane complex of gram-negative microorganisms. Its causative role in gram-negative bacteria-induced diseases and broad applications in different kinds of cell stimulation experiments provided a conceptual basis for studies directed at the isolation, purification, and detailed chemical characterization of LPS. The main problem with LPS purification protocols is the contamination of the end product with nucleic acids and proteins in variable proportions which could potentially interfere with downstream applications. In this study, a simple procedure for purification of LPS from Escherichia coli (E. coli) and Salmonella typhi (S. typhi) with high purity and very low contaminating nucleic acids and proteins based on the hot phenol-water extraction protocol has been introduced. The purity of extracted LPS was evaluated by silver and coomassie blue staining of SDS-PAGE gels and HPLC analysis. Limulus Amebocyte Lysate (LAL) coagulation activity and rabbit pyrogen assay were exploited to monitor the functionality of purified LPS. The results showed that DNase and RNase treatment of the sample is essential after the sonication step to eliminate nucleic acid contamination in the LPS fraction. Silver staining demonstrated ladder pattern which is characteristic of LPS. No contaminating protein was found as assessed by Coomassie blue staining. HPLC fractionation revealed high degree of purity comparable with commercial LPS. Parenteral administration of purified LPS resulted in substantial increase of rabbits’ body temperature (mean: 1.450C). LAL coagulation assay confirmed the functional activity of the purified LPS. In conclusion, the protocol presented here could be employed for isolation of LPS with high purity and functional activity.
Introduction :
Lipopolysaccharide (LPS) is the main outer membrane component of gram-negative bacteria which constitutes about 75% of the surface (1) and 5-10% of the total dry weight of gram-negative bacteria (2). Their basic structure consists of three parts: lipid A, core oligosaccharide and repetitive polysaccharide designated as O antigen. Lipid A is highly conserved and exerts the endotoxic activity, while the O antigen carbohydrate chain is a polymer of repeating oligosaccharides, which differs between species and is responsible for the serological specificity of bacteria (3). LPS causes pathophysiological effects such as fever, leucopenia, leucocytosis and Shwartzman reactivity (4,5).
The recognition of very significant causative role of LPS in gram negative bacteria-induced diseases prompted many researchers to conduct studies directed at its isolation and purification. Therefore, it is not surprising that a plenty of methods and protocols have been introduced for the isolation and purification of LPS from bacteria included among them are trichloroacetic acid extraction at 4 ºC (6), aqueous butanol (7), triton/Mg+2 (8), cold ethanol (9) and extraction in water at 100 ºC (10). Other purification protocols with phenol, chloroform, petroleum-ether (11) and methanol (12) have been described specifically for rough LPS.
Recently a combination of Westphal method based on the hot phenol extraction procedure and sized exclusion chromatography was successfully employed for the purification of LPS from E.coli (13). The method proposed by Westphal (14) is still the most frequent procedure employed for LPS extraction because of its high yield. Contamination with proteins and nucleic acids are among the main disadvantages of some proposed protocols for LPS purification which hinder reliable application of the end product in such sensitive assays as molecular and immunological experiments. Although ultracentrifugation can be employed for elimination of contaminating proteins (7,14), this usually leads to lower yields and considerable amount of nucleic acids contaminate the sedimented LPS (14).
In this view, no single method is suited to isolation of LPS with high purity and combination of two purification steps maybe necessary. In the present study, we have employed a modified phenol-water extraction protocol accompanied with proteinase K digestion of bacterial proteins and nuclease elimination of nucleic acids for extraction of LPS from E. coli and S. typhi with high purity.
Materials and Methods :
Bacterial strains and growth conditions
E. coli 055:B5 (Pasteur Institute, Paris, France) and Salmonella typhi B-34-2 (Pasteur Institute, Tehran, Iran) were grown in Luria-Bertani broth medium (usb, Cleveland, USA) at 37 °C in shaker incubator overnight. After centrifugation of culture media, sedimented bacteria were harvested and used for LPS extraction and purification.
LPS extraction and purification
LPS was extracted by hot phenol-water method as described previously with some modifications (14). In brief, bacterial suspensions (108 colony-forming units/mL) were centrifuged at 10,000 × g for 5 min. The pellets were washed twice in PBS (pH=7.2) (0.15 M) containing 0.15 mM CaCl2 and 0.5 mM MgCl2. Pellets were then resuspended in 10 ml PBS and sonicated for 10 min on ice.
In order to eliminate contaminating protein and nucleic acids, treatment with proteinase K, DNase and RNase was performed prior to extraction step. For this purpose, proteinase K (100 μg/mL) (Roche, Mannheim, Germany) was added to the cell mixture and the tubes were kept at 65 °C for an additional hour. Mixture was subsequently treated with RNase (40 μg/mL) (Roche, Mannheim, Germany) and DNase (20 μg/mL) (Roche, Mannheim, Germany) in the presence of 1 μL/mL 20% MgSO4 and 4 μL/mL chloroform and incubation was continued at 37 C overnight.
At the next step, an equal volume of hot (65–70 °C) 90% phenol was added to the mixtures followed by vigorous shaking at 65-70 °C for 15 min. Suspensions were then cooled on ice, transferred to 1.5 mL polypropylene tubes and centrifuged at 8500 × g for 15 min. Supernatants were transferred to 15-mL conical centrifuge tubes and phenol phases were reextracted by 300 μL distilled water. Sodium acetate at 0.5 M final concentration and 10 volumes of 95% ethanol were added to the extracts and samples were stored at –20 °C overnight in order to precipitate LPS.
Tubes were then centrifuged at 2000 × g 4 °C for 10 min and the pellets were resuspended in 1 ml distilled water. Extensive dialysis against double distilled water at 4 °C was carried out at the next step until the residual phenol in the aqueous phases was totally eliminated. Final purified LPS product was lyophilized and stored at 4 °C.
Silver, commassie blue and ethidium bromide stainings
The purified LPS was solubilized in sample buffer to the desired concentration (1 mg/mL), and boiled for 5 min. 16 μL/well from each sample was separated on 15% SDS gel with a 4% stacking gel under reducing condition at 100 mA for 2 hr using mini-PROTEAN electrophoresis instrument (Bio-Rad Laboratories, California, USA). Silver and coomassie blue staining of the gels was performed according to the standard protocol. Staining of agarose gel with ethidium bromide was done as well in order to show if any contamination with nucleic acids persist. To do this, 10 μL/well of reconstituted LPS from both E. coli and S. typhi and also 10 μL/well bacterial suspensions (108 colony-forming units/mL), as positive control, were loaded on agarose gel and stained with ethidium bromide.
High performance liquid chromatography (HPLC)
HPLC separations were carried out by a Knauer Smartline 1000 pump equipped with a Smartline UV detector 2500 (Berlin, Germany), and a Rheodyne 7725 injection valve (Cotati, CA, USA). The method was optimized at the 0.8 ml/min flow rate and 210 nm wavelength UV detection. Separation was carried out over C18 column with 4.6 mm diameter and 250 mm length from Grace Company (Munich, Germany) with a mixture of water and acetonitrile (95:5) as mobile phase. Extra pure E. coli LPS (Sigma, Saint Louis, USA) was used as standard.
Limulus amebocyte lysate (LAL) assay
The potency of LPS samples were determined by the limulus amebocyte assay gel-clot method (LONZA, Walkersville, USA) which had a sensitivity of 0.06 endotoxin units per milliliter (UE/mL), according to the protocol published elsewhere (15).
Rabbit pyrogen test
Two white New Zealand rabbits weighing between 1.7 and 2.3 kg (Pasture Institute, Tehran, Iran) were used. Pyrogenicity was tested in an air conditioned room. Rabbits were given an intravenous injection of 5 ng/kg of body weight of purified LPS. Rectal temperatures were measured with indwelling rectal thermostats and recorded before injection and 4 hr after pyrogen administration. One rabbit was injected with sterile PBS as control.
Result :
SDS-PAGE separation and silver and coomassie blue staining of LPS
Separation over SDS-PAGE gel followed by silver staining was used to detect and visually characterize the purified LPS. Sliver staining is a highly sensitive method capable of detecting as low as 1 ng LPS and is routinely used for visualization of the band pattern of purified LPS.
As depicted in figure 1A, LPS from both E. coli and S. typhi gave a characteristic dark staircase (ladder-like) pattern of bands. Lipid A-core LPS migrating very near the dye-front stained very intensely and appeared as black region at the bottom of gel. The results also showed different profile of LPS banding in the two strains studied. Coomassie blue staining of the gels showed no band indicating absence of contaminating proteins (Figure 1B). Staining of agarose gel with ethidium bromide and absence of any band in purified LPS products showed no contamination with nucleic acid (Figure 1C).
HPLC analysis of purified LPS
The purity of LPS isolated from E. coli and S. typhi was assessed by HPLC. The band profile of LPS extracted from E. coli was analyzed and compared to that of extra pure commercial standard. As shown in figure 2, the LPS chromatogram of purified LPS from E. coli overlaid with that of standard indicating high purity of the product. Chromatogram of purified S. typhi LPS also showed a single major sharp band suggesting very low content of impurities.
Assessment of LPS functional activity by LAL test
The principle of LAL coagulation assay is based on the fact that endotoxin activates the Limulus Amoebocyte Lysate (LAL) pro-enzyme, resulting in gel formation. The result of our qualitative LAL coagulation assay indicated the functional activity of purified LPS as demonstrated by the formation of gel in vials containing the LAL.
Evaluation of purified LPS pyrogen activity by rabbit pyrogen test
The endogenous pyrogen activity of purified LPSs was evaluated using rabbit pyrogen test. The initial rectal temperatures of the two rabbits were 38.3 °C and 38.4 °C. Injection of LPS from E. coli and S. typhi, caused raising the rectal temperature up to 39.7 °C and 39.9 °C respectively. The control rabbit which received PBS did not show considerable fluctuation of body temperature.
Discussion :
LPS is the main component of cell membrane of almost all gram negative bacteria. It is responsible for the pathological consequences of gram negative bacterial infections. Such life threatening diseases as septic shock following infection with gram negative bacteria is mediated mainly by LPS. LPS is potent activator of immune system capable of triggering cytokine release from cells of different origin. It is widely used as an inducer of TLR-4 signaling pathway (16) and as a mediator of dendritic cell maturation (17).
Conclusion :
Although the purity of LPS is a good measure of the performance of purification system, functional activity of the final product is important as well. In this context, the results of rabbit pyrogen and LAL coagulation tests clearly proved the functional activity of the purified product. In conclusion, the protocol presented here could be employed for isolation of LPS with high purity and functional activity from different strains of smooth gram negative bacteria which have structurally different LPS.
Acknowledgement :
This work was supported financially by Avicenna Research Institute, and Tehran University of Medical Sciences.
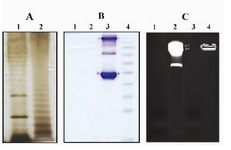
Figure 1. Silver, coomassie blue and ethidum bromide stainings of purified LPS
LPS from E coli (Lanes 1A and 1B) and S.typhi (lanes 2A and 2B) was purified by modified hot phenol-water extrac-tion method and fractionated by SDS-PAGE electrophore-sis followed by silver (A) or commassie blue staining (B). Ladder pattern of LPS banding which is charasteristic of smooth gram negative bacteria is seen (A). The absence of band in commassie blue staining as shown in B indicates no contamination of purified LPS with bacterial proteins. Lane 3B: Human IgG and BSA, Lane 4B: Molecular weight marker. Residual nucleic acid contamination in purified LPS products was traced by eithidium bromide staining (C). Absence of band in LPS from E.coli (Lane 1C) and S.typhi (Lane 3C) shows no contamination with nucleic acids in purified LPS products. Lane 2C and 4C: Whole E. coli and S.typhi, respectively
|
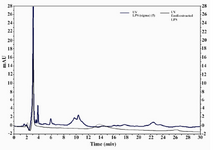
Figure 2. HPLC fractionation of LPS from E.coli. The gray and blue lines show chromatograms of purified E.coli LPS and extra pure commercial LPS from E.coli, respectively
|
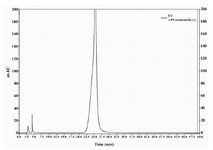
Figure 3. HPLC chromatogram of LPS extracted from S. typhi
|
|