Modern Paradigm Towards Potential Target Identification for Antiviral (SARS-nCoV-2) and Anticancer Lipopeptides: A Pharmacophore-Based Approach
-
Yadav, Manisha
-
Department of Biotechnology, National Institute of Technology Raipur, Chhattisgarh, India
-
 Eswari, J. Satya
Department of Biotechnology, National Institute of Technology Raipur, Chhattisgarh, 492010, India, E-mail: satyaeswarij.bt@nitrr.ac.in
Eswari, J. Satya
Department of Biotechnology, National Institute of Technology Raipur, Chhattisgarh, 492010, India, E-mail: satyaeswarij.bt@nitrr.ac.in
Abstract: Background: Lipopeptides are potential microbial metabolites that are abandoned with broad spectrum biopharmaceutical properties ranging from antimicrobial, antiviral and anticancer, etc. Clinical studies are not much explored beyond the experimental methods to understand drug mechanisms on target proteins at the molecular level for large molecules. Due to the less available studies on potential target proteins of lipopeptide based drugs, their potential inhibitory role for more obvious treatment on disease have not been explored in the direction of lead optimization. However, Computational approaches need to be utilized to explore drug discovery aspects on lipopeptide based drugs, which are time saving and cost-effective techniques.
Methods: Here a ligand-based drug discovery approach is coupled with reverse pharmacophore-mapping for the prediction of potential targets for antiviral (SARS-nCoV-2) and anticancer lipopeptides. Web-based servers PharmMapper and SwissTargetPrediction are used for the identification of target proteins for lipopeptides surfactin and iturin produced by Bacillus subtilis.
Results: The studies have given the insight to treat the diseases with next-generation large molecule therapeutics. Results also indicate the affinity for Angiotensin-Converting Enzymes (ACE) and proteases as the potential viral targets for these categories of peptide therapeutics. A target protein for the Human Papilloma Virus (HPV) has also been mapped.
Conclusion: The work will further help in exploring computer-aided drug designing of novel compounds with greater efficiency where the structure of the target proteins and lead compounds are known.
Introduction :
The focus of the current trends in computer-aided drug discovery is to understand the disease mechanism, and further target identification takes place. On the edge of personalized medicine, the system is based upon the molecular basis of fundamentals in drug designing 1. The interplay mentioned above can be addressed by imparting the cheminformatics tools in high throughput format. Cancer is a life-threatening disease which is one of the severe health problem globally. The increasing drug resistance has urged for the search of novel anticancer agents 2. However, steadily increasing drug resistance in the treatment of infectious disease posed a severe problem in antimicrobial therapy and necessitates continuing research on different classes of drug derivatives 3.
Lipopeptides are known as anti-microbial, anti-tumor, anti-inflammatory, anti-hypertensive, anti-parasitic, and anti-cancer compounds 4. The lipopeptides exhibit various biological activities due to the presence of lipid and peptide moiety. Moreover, numerous therapeutically important medicines like peptide therapeutics contain a heterocyclic nucleus. Lipopeptides are amphiphilic compounds of natural origin and found with various therapeutic properties. Surfactin and Iturin lipopeptides produced by Bacillus subtilis (B. subtilis) are reported with antiviral and anticancer properties 5-8. The studies on lipopeptides have been explored for antiviral activities against Corona virus (SARS-nCoV-2) 9. Large molecule therapeutics are found with higher target specificity as compared to small molecules and has the potential to inhibit target proteins from multiple sites figure 1. Hence exploring the drugs with higher selectivity help in subsiding side effects and non-specific cytotoxicity of small-molecule
inhibitors in cancer treatment 10,11.
The first and subsequent second wave of COVID-19 pandemic due to the outbreak of SARS-nCov-2 (South East Respiratory Syndrome-Novel Corona Virus-2) is concurring the world leading to the global emergency to seek potential treatment options. The current management of the disease through drug repurposing has been an efficient way to combat the corona virus infection. Various target proteins and potential drug options are being explored to defeat the viral pathogenicity 12. Lipopeptides have been known as potential antiviral compounds and such therapeutic options are also being researched 13,14. The need of the hour is to first map the viral targets with the available potential therapeutics in an order to understand their mechanism of action for the novel drug discovery and drug repurposing 12,15. Such studies will facilitate to further accelerate the drug discovery pipeline to combat future pandemics through the discovery of next generation therapeutic options such as peptides and lipopeptides.
There is an urgent need to explore such a new class of drugs where the drug resistance is threatening the world with a slow pipeline of drugs instead of having fledged with increased knowledge and technology-driven facilities 10. The struggle starts with the identification of potential targets for large molecule therapeutics 16. Here a drug discovery approach is coupled with pharmacophore-based virtual screening for the effective prediction of potential targets for lipopeptides. The studies have not been explored in the direction of lead optimization of natural compounds to increase the target specificity of the drug. Though, target identification is the primary step towards novel drug discovery. In silico methods, facilitate the cost-effective ways for target identification and lead optimization in reduced time and chemical exposure 7. Structure-based pharmacophore mapping is a useful technique when insufficient information on ligand molecule is available and therapeutic activity (induce or block) for a particular disease is experimentally proved. It is used to explore information about the receptor site. It gives more in-depth insight for the receptor-ligand interaction at the molecular level 17.
In view of the above mentioned facts, compounds derived from synthesis are directed towards the proteomic approach extensively for the identification of potential binding proteins. These techniques are based upon comparative studies on protein expression profiling concerning the presence and absence of a given molecule for a particular cell or tissue. Such methods are not much successful in the discovery of target proteins because these are time consuming and laborious 18. In the case of large molecule natural compounds, the task is even more challenging to execute and incurs huge wastage of chemicals and time 19. Therefore, to bring such natural compounds into the drug discovery pipeline, computational approaches followed by advanced synthetic techniques lead to more significant benefits to improve the health and well-being with the help of next generation drugs 6,20. In the current work in-silico method for target profiling is implemented for the identification of target proteins responsible for cancer and viral infections such as SARS-nCov-2 against anticancer and antiviral lipopeptides surfactin and iturin using web-based servers.
Materials and Methods :
Pharmacophore Mapping using PharmMapper: Reverse pharmacophore mapping approach is used with the help of a web-based server PharmMapper. The studies are conducted for identification of target proteins for lipopeptide compounds with anticancer and antiviral properties 4,21. The techniques have been previously utilized for the target proteins for essential oils of Cardomom and bis-pyrimidine compounds 22. The PharmMapper server utilizes the reverse pharmacophore mapping to identify the potential targets for a given query compound. It utilizes an in-built database of pharmacophore models of 23,236 annotated proteins from targetBank, BindingDB, DrugBank, and Potential Drug Target Database (PDTD) with 51,431 ligandab-ble and 16,159 druggable pharmacophore models. The server compares the query compound with models of the in-built database.
According to the similarity of the pharmacophore of the query compound with identified pharmacophore of the target proteins, the results are provided in the form of Zscore. Alongside the importance and indications of the identified protein in the disease are also provided 23. Here the potential anticancer peptidolipidic compounds of B. subtilis are submitted as the query compound for possible target identification 24. The selection of target proteins is done based on their importance in the cancer disease. The selected lipopeptides are reported with anticancer properties in literature.
The query compounds were submitted to PharmMapper server (http://59.78.96.61/pharmmapper). The PharmMapper server compares the pharmacophore of the query compound with an in-built database of pharmacophore models. It predicts 300 target proteins based on their fitness score and pharmacophoric features. The target protein was ranked as per the fitness score and importance and indication of the protein. Generally, the top 10 proteins with a fitness score of more than 5 were considered to identify the probable target proteins of the query compound 18.
Swiss Target Prediction for target identification: Swiss Target prediction is a web-based server developed by the Swiss Institute of Bioinformatics. This tool is used for the prediction of potential targets of a compound for which targets with Protein ID has not been explored. The website facilitates the estimation of probable targets of a query compound or a bioactive compound. The server has an in-built library of 370,000 known active compounds and more than 3000 target proteins from various species. In the updated version of the server, a large dataset on drug-protein interaction has been made available, which makes it a unique source of information. It is based upon a knowledge-based development approach for the identification of novel targets or secondary targets for uncharacterized compounds or known compounds, respectively. The server accurately predicts the targets for bioactive compounds based on two-dimensional and three-dimensional (2D and 3D) measures of similarity respective to the known compounds 25-27.
Lipopeptides are known as potential anti-cancer, anti-viral compounds. However, the approach towards the identification of the target protein has not been explored. Hence, computational methods for pharmacophore mapping are utilised, which facilitate the identification of potential targets for lipopeptide-based drugs.
Selection and retrieval of lipopeptides: Lipopeptides with potent anticancer properties have been identified and screened through the rigorous literature survey. Surfactin and iturin have been chosen as the most prominent anticancer lipopeptides. The PubChem compound database is utilized to get the compound information. The canonical SMILES annotation of the query compounds were used to submit the molecules to the server and can be used to draw the compound into two-dimensional structure. The information were submitted into the servers in the form of canonical SMILES for query compounds.
Pharmacophore mapping: The two-dimensional structures of the above-mentioned lipopeptides were drawn using the information of canonical SMILES from the PubChem compound database., The 2D sketcher tool of Maestro suite (Schrodinger software package) was used to draw the structures. The server utilizes the sdf and mol2 (mole file formats/extensions for saving chemical structures) file formats for the submission of the query compound for pharmacophore mapping. Further, the individual structures were submitted to the software PharmMapper, which is a free web-based tool for the identification of target proteins based upon the structural complementarity with the submitted drugs. Results obtained through Pharmacophore mapping were further subjected to the structure prediction of the given proteins. Software is capable of providing a minimum of 10-300 target proteins based on their binding vicinity in which the drug should be able to dock and inhibit the target protein. The traditional sequence alignment methodology followed by structure prediction, and further data mining approaches were used for obtaining the three-dimensional structures of predicted proteins.
Sequence alignment & structure prediction: This step was performed using the retrieval of the FASTA sequence of each protein given in the mapping results. The FASTA sequence has further proceeded for sequence alignment using the Basic Local Alignment Search Tool abbreviated as BLAST, a web-based algorithm provided by the National Centre for Biotechnology Information (NCBI). On the basis of alignment score and keeping the query coverage in consideration, the higher similarity structures were retrieved from the Protein DataBank (PDB). The PDB structures were used as a template for the homology modeling of the FASTA sequence of the target proteins predicted through PharmMapper. Some of the FASTA sequences were traced with the 100% similarity in the sequence alignment. However, the experimental structures are available in the protein databank. Hence the structures can be retrieved for further studies.
Target identification: In silico target identification for drug molecules is the primary step in drug discovery, which includes various algorithms for the identification of genes and proteins. The availability of 3D structure of the target protein enables the identification of the best binding mode for understanding the interaction. Pharmacophore is a spatial arrangement of functional groups, is an essential core of the molecule which interacts with the receptor target molecule is the alternative method for deciphering molecular interaction.
Multiple sequence alignment using T-Coffee server: As per pharmacophore mapping results, the mapped protein with PDB ID 2R5K major capsid protein L1 of HPV (Human Papilloma Virus) (Table 1) was further subjected to multiple sequence alignment with SARS-nCoV-2 nucleocapsid protein. The FASTA sequences of both the proteins were retrieved from NCBI and submitted to the T-Coffee server, which uses Clustal_W algorithm for multiple sequence alignment. Multiple sequence alignment is done to have an insight into the percentage similarity index between these two sequences. Sequence similarity searching helps in the identification of feasibility of drug repurposing for a given compound, as established antiviral drugs are repurposed to treat infection of SARS-nCoV-2 which is responsible for current COVID-19 pandemic.
FASTA sequences of Major Capsid protein L1 of HPV:
>pdb|2R5H|D Chain D, Late Major Capsid Protein L1
AVVSTDEYVARTNIYYHAGTSRLLAVGHPYFPIKKPNNNKILVPKVSGLQYRVFRIHLPDPNKFGFPDTS
FYNPDTQRLVWACVGVEVGRGQPLGVGISGHPLLNKLDDTENASAYAANAGVDNRECISMDYKQTQLCLI
GCKPPIGEHWGKGSPCTQVAVQPGDCPPLELINTVIQDGDMVDTGFGAMDFTTLQANKSEVPLDICTSIC
KYPDYIKMVSEPYGDSLFFYLRREQMFVRHLFNRAGTVGENVPDDLYIKGSGSTANLASSNYFPTPSGSM
VTSDAQIFNKPYWLQRAQGHNNGICWGNQLFVTVVDTTRSTNMSLCAAISTSETTYKNTNFKEYLRHGEE
YDLQFIFQLCKITLTADVMTYIHSMNSTILEDWNGGSGGEDPLKKYTFWEVNLKEKFSADLDQFPLGRKF
LLQL
FASTA sequence of Nucleocapsid phosphoprotein of SARS-nCoV-2:
>QHR84456.1 nucleocapsid phosphoprotein [Severe acute respiratory syndrome coronavirus 2]
MSDNGPQNQRNAPRITFGGPSDSTGSNQNGERSGARSKQRRPQGLPNNTASWFTALTQHGKEDLKFPRGQ
GVPINTNSSPDDQIGYYRRATRRIRGGDGKMKDLSPRWYFYYLGTGPEAGLPYGANKDGIIWVATEGALN
TPKDHIGTRNPANNAAIVLQLPQGTTLPKGFYAEGSRGGSQASSRSSSRSRNSSRNSTPGSSRGTSPARM
AGNGGDAALALLLLDRLNQLESKMSGKGQQQQGQTVTKKSAAEASKKPRQKRTATKAYNVTQAFGRRGPE
QTQGNFGDQELIRQGTDYKHWPQIAQFAPSASAFFGMSRIGMEVTPSGTWLTYTGAIKLDDKDPNFKDQV
ILLNKHIDAYKTFPPTEPKKDKKKKADETQALPQRQKKQQTVTLLPAADLDDFSKQLQQSMSSADSTQA.
Results and Discussion :
Prediction of target proteins for anticancer activities of lipopeptides: PharmMapper and Swiss Target Prediction predict the target proteins for a ligand molecule on the basis of the vicinity of the binding cavity of the target proteins, which can probably be inhibited through the query ligand molecule. The query ligand was used here is lipopeptide surfactin (Figure 2) for which the possible target proteins have been predicted and given in the table 1. According to the PharmMapper report depicted in (Table 1), hemoglobin subunits and membrane proteins have been predicted, which are associated with the hemolytic and surface active properties of surfactin, respectively 28.
As per the PharmMapper results depicted in the (Table 1), top 10 ranked protein IDs are considered as the most significant target proteins. Resulting protein with PDB ID 4YU4 is a hemoglobin subunit that significantly proved to be associated with the hemolytic properties of the surfactin. As per the experimental studies, the hemolytic property is a hinderence of using surfactin as a potential anticancer compound. This was never been explored at the molecular level to understand the factors involved in the hemolytic property of the lipopeptide-based compounds 29. Another protein with PDB ID predicted is 3CK1, a membrane protein which suggests the potential membrane affinity of the compound to lead its pore-forming activity as an antimicrobial compound. As FDA approved lipopeptide daptomycin is successfully used to treat the infections of multidrug-resistant bacteria 30. To explore the anticancer activity of compounds, it is necessary to understand the interaction of the ligand at the target site. This study will give an insight into the lead optimization of the surfactin molecule to identify novel analogs with low or non-haemolytic property and higher affinity towards the target proteins for cancer and viral diseases. The studies would be a stepping stone towards the discovery of next-generation anti-cancer and anti-viral compounds 24,31,32.
As per Swiss Target Prediction report in figure 3 and table 2, major proteins predicted are from the apoptotic pathway, which is having significant role in the cancer progression. Inhibition of such a signaling pathway plays a major role in cell death, which has been explained in the mechanism of action of anti-cancer lipopeptides that they induce the cell cycle arrest and leads to cell death 33. The predicted proteins can be further explored with the help of structural bioinformatics and computer-aided drug designing studies. The work can be extended further for lead optimization studies for a given class of drugs to design a molecule with better efficiency and specificity to inhibit a particular target protein 7,34.
The Swiss Target Prediction report in figure 3 is depicting the surfactin structure as a query compound, and a pie chart is depicting the percentage mapping with the target class. The results show surfactin as a potential ligand for major proteases (60%), adhesins, and membrane receptors. Lipopeptides being amphiphilic compounds are found with membrane affinity properties, which can be proved with these results 35,36. Tables 2 and 3 suggest the Caspase proteins as the potential class of target proteins for surfactin. Caspases have a significant role in cancer progression 37. Being a potential target of peptide therapeutics, this aspect can be explored in conjunction with inhibitory/catalytic activity to regularize the class of caspase proteins in cancer management 10,37.
Swiss Target Prediction report generated against lipopeptide iturin reveals the G-Protein Coupled Receptors (GPCR) as the most prominent class of targets for iturin (Figure 4 and Table 4). Drugs targeting GPCR’s are used to treat many diseases. More than 40% FDA approved drugs are aimed to target GPCR’s and their related pathways. Now, GPCR’s are being used as biomarkers for early diagnosis of cancer, as they play critical role in activating and regulating signaling pathways associated to cancer 38. Hence, iturin would be a promising compound to be explored to develop as potential anticancer drug 38.
Prediction of viral targets for antiviral activity of lipopeptides: Pharmacophore mapping studies suggested various viral targets along with the anticancer studies. The studies contain significant findings for the identification of next-generation antiviral drugs to combat the diseases like COVID-19. As per table 1 multidrug resistance operon repressor is found as a predicted target, which suggests the proven antibiotic activity against multidrug resistance strains 6. Table 4 depicts the Angiotensin-Converting Enzyme (ACE) as one of the potential targets of iturin. The affinity of a compound towards ACE2 is significant as Human-ACE2 is a potential target for SARS-nCov-2 Spike-glycoprotein. Inhibition of such interaction plays a vital role in the treatment of Corona virus disease (COVID-19) 9. The findings from table 1 generated through PharmMapper results for surfactin, suggested the affinity towards major capsid protein of Human Papilloma virus (PDB ID: 2R5K). Clustal_W analysis results gave approximately 21% similarity with SARS-nCoV-2 nucleocapsid protein. The results suggest the drug repurposing opportunity with the identification of a similar category of drugs with a high affinity towards SARS-nCoV-2 viral proteins 15,39. Table 4 is also suggesting affinity towards a viral protein Baculoviral IAP repeat-containing protein 40.
Conclusion :
Synergistically conducted studies on target protein identification of microbial peptidolipidic compounds give more promising and reliable results regarding the target class of compounds. The experimental studies were touched at the level of mechanistic action but could not lead towards optimization and novel analog designing, which required the structural and molecular information of drug and target protein. Through the in silico studies, the multitargeting efficiency of lipopeptide compounds is depicted, which is not feasible with a small molecule inhibitor. This suggests the higher specificity and efficiency in binding with the target site. In the current studies, antiviral and anticancer properties of lipopeptide compounds have given various horizons for future perspectives in developing next-generation novel compounds. It is the need of the hour to explore novel compounds to combat future infectious diseases like COVID-19 pandemic and deadly cancer diseases. The study concludes various classes of potential viral targets and cancer target proteins to facilitate the drug discovery process with the possible targets and ligands to study the interaction and discover the promising drug candidates. Utilization of microbial bi-products is much feasible than producing peptide therapeutics in the laboratory, which incurs huge chemical exposure and a financial burden as well with the wastage of chemicals, time and money. Rather computational prediction eases out the journey to some extent. It gives a better understanding of the drug-target interaction at the molecular level. The research can be further explored with molecular dynamics studies for a more in-depth understanding of lead optimization and exploring these compounds with more potency and least side effects.
Acknowledgement :
We are thankful to the National Institute of Technology Raipur and Chhattisgarh Council of Science and Technology (CCOST) (Project number 2487/ CCOST/MRP/2016, Raipur dated 25.01.2016), India for providing the necessary facilities to prepare the manuscript and permission to publish it.
Conflict of Interest :
The authors declare no conflict of interest.
Ethical Approval :
This article does not contain any studies with human participants or animals performed by any of the authors
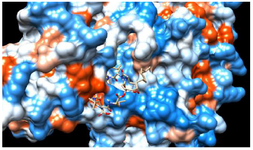
Figure 1. Molecular docking interaction: cyclic peptide drug docked with target protein (electrostatic surface interaction). Ability of cyclic peptide to inhibit target from multiple sites.
|
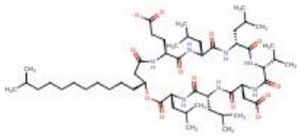
Figure 2. 2D structure of cyclic lipopeptide–surfactin (query compound).
|
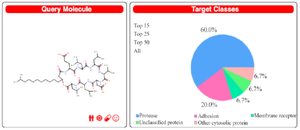
Figure 3. Swiss Target prediction query structure for Surfactin and prediction report with percentage affinity with class of target protein.
|
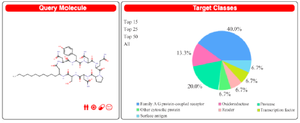
Figure 4. Iturin as query molecule for Swiss Target Prediction with prediction report in percentage for target class.
|
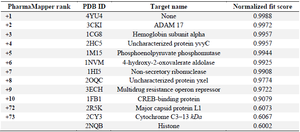
Table 1. PharmMapper report for Surfactin: Depicting Hemoglobin subunit, multidrug-resistant operon, Major capsid viral protein and Histones as potential target class
|
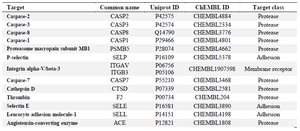
Table 2. Surfactin Swiss Target Prediction report depicting Caspases, Proteases and Membrane receptor proteins as potential targets classes and deciphering Angiotensin Converting enzyme (ACE2) as potential target against SARS-nCoV-2 under top 50 targets aligned
|
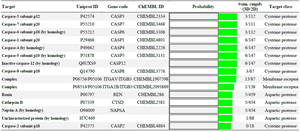
Table 3. SwissTarget Prediction report for Surfactin
|
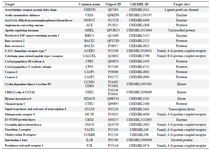
Table 4. Swiss Target Report (prediction of GPCR’s, Caspases and ACE as targets for Iturin) and predicted Cyclin dependent kinases (CDK’s) through mapping against Iturin
|
|