Genome Analysis of the Enterococcus faecium Entfac.YE Prophage
-
Elahi, Yara
-
Department of Genetics, Faculty of Life Sciences, Islamic Azad University Tehran North Branch, Tehran, Iran
-
Seifi, Arash
-
Department of Infectious Diseases, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran
-
Mahfouzi, Saeideh
-
Department of Virology, School of Public Health, Tehran University of Medical Sciences,, Tehran, Iran
-
 Saboor Yaraghi, Ali Akbar
Department of Pathobiology, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran, Tel: +98 21 42933168; E-mail: asaboor@tums.ac.ir
Saboor Yaraghi, Ali Akbar
Department of Pathobiology, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran, Tel: +98 21 42933168; E-mail: asaboor@tums.ac.ir
Abstract: Background: Bacteriophages are viruses that infect bacteria. Bacteriophages are widely distributed in various environments. The prevalence of bacteriophages in water sources, especially wastewaters, is naturally high. These viruses affect evolution of most bacterial species. Bacteriophages are able to integrate their genomes into the chromosomes of their hosts as prophages and hence transfer resistance genes to the bacterial genomes. Enterococci are commensal bacteria that show high resistance to common antibiotics. For example, prevalence of vancomycin-resistant enterococci has increased within the last decades.
Methods: Enterococcal isolates were isolated from clinical samples and morphological, phenotypical, biochemical, and molecular methods were used to identify and confirm their identity. Bacteriophages extracted from water sources were then applied to isolated Enterococcus faecium (E. faecium). In the next step, the bacterial genome was completely sequenced and the existing prophage genome in the bacterial genome was analyzed.
Results: In this study, E. faecium EntfacYE was isolated from a clinical sample. The EntfacYE genome was analyzed and 88 prophage genes were identified. The prophage content included four housekeeping genes, 29 genes in the group of genes related to replication and regulation, 25 genes in the group of genes related to structure and packaging, and four genes belonging to the group of genes associated with lysis. Moreover, 26 genes were identified with unknown functions.
Conclusion: In conclusion, genome analysis of prophages can lead to a better understanding of their roles in the rapid evolution of bacteria.
Introduction :
Two important species of commensal enterococci, Enterococcus faecalis (E. faecalis) and Enterococcus faecium (E. faecium), are one of the leading causes of medical conditions, causing various hospital infections such as endocarditis and sepsis 1. Due to the increased antibiotic resistance properties of these bacteria, their infections are often difficult to treat 2. For example, the prevalence of Vancomycin-Resistant Enterococci (VRE) has increased inducing complexities in hospitalized patients in the last two decades 3. Since one of the most important health concerns is to find novel solutions to fight these multidrug-resistant bacterial infections, the use of novel strategies seems urgently necessary. In this regard, the use of phages can hopefully be promising 4. Bacteriophages (Phages) are prokaryotic viruses detected in various environments within their bacterial hosts or in large numbers of free virions 5. Currently, these bacterial viruses are under the spotlight as appropriate substitutes for the available antibiotics. Phages can effectively infect and kill antibiotic-resistant bacteria regardless of the resistance patterns of these bacteria 6. In addition, phages have several advantages over other antimicrobial agents with no serious or irreversible side effects 7. Therefore, the characterization of novel phages and understanding their evolutionary ecology can greatly help scientists improve the process of chemical antimicrobial replacement. Novel genome analyzing methods, including next-generation sequencing methods, and established genome databases have relatively facilitated the development of phage knowledge. Therefore, the purpose of the current study was to analyze the Entfac.YE prophage.
Materials and Methods :
Bacterial strain: E. faecium was isolated from clinical samples in a university teaching hospital in Tehran, Iran, using routine culture methods as well as phenotypic and genotypic methods 8. The isolate was verified using morphological, biochemical, and molecular techniques such as catalase test, arabinose fermentation, salt tolerance, optochin susceptibility, CAMP, and PYR tests. Enterococcal tuf gene was amplified using PCR (Polymerase chain reaction) and partially sequenced using the Sanger sequencing platform (Kawsar Biotech, Iran). The bacterial strain was also used for the isolation of enterococcal phages.
Whole-genome sequencing: After isolation of phages on E. faecium, the bacterial strains challenged by the phages were used to extract their genome. Briefly, a small volume of two-layer agar-containing bacteria with phages was removed and dissolved in Saline Magnesium (SM) buffer. This was centrifuged at 4480 g for 10 min. After centrifugation, the supernatant was filtered through 0.45-μm syringe filters and mixed with DNase 1 and RNase A. The mixture was stored at 37°C for 30 min. Then, the bacterial genome was extracted using the precipitation method with ethanol and propanol. The extracted bacterial genome was completely sequenced using the Illumina Hiseq platform (Novogene, China) (Table 1). SPAdes algorithm was used in the de novo technology of genome assembling. Furthermore, the reference assembly method was used for the raw data. The prophage was analyzed using Regulatory Sequence Analysis Tools (RAST) (https://rast.nmpdr.org/) and then DDBJ Nucleotide Sequence Submission System (NSSS) (https://www.ddbj.nig.ac.jp).
Results :
Sanger sequencing results of the bacterial tuf gene verified the initial characteristics of the isolated bacteria (DDBJ accession numbers: LC580430 and LC580431). The bacterial genome was completely sequenced and information obtained through Novaseq 6000 platform (Illumina, USA) are demonstrated in table 1. The Entfac.YE prophage included 69,990 nucleotides, consisting of 31.13% A, 31.60% T, 18.94% C, and 18.29% G nucleotides. In total, 88 prophage genes were analyzed for their functions (Table 2). The prophage content included four housekeeping genes, 29 genes in the group of replication and regulation, 25 genes in the group of structure and packaging, and four genes in the group of lysis. The functions of other 26 genes were unknown (Figure 1). Bacteriophages isolated in a relative phase of the current study included three lytic members. Two tailed phages included isometric shapes (Siphoviridae and Myoviridae) and the other one was filamentous (Inoviridae).
Discussion :
In general, phages isolated from the clinical strain of E. faecium using sources of wastewaters were lysogenic phages with the suggestion of further prophages in the bacterial genome. Therefore, whole-genome sequencing of the enterococcal strain was carried out. The selected prophage of E. faecium was named Entfac.YE, including 88 genes. Of the identified genes, four genes were in the group of housekeeping genes, 29 genes were in the group of replication and regulation genes, 25 genes were in the group of packaging and structure genes, four genes were in the group of lysis genes, and 26 genes were in no groups due to their unknown functions. In this study, 14 E. faecium were identified in 25 enterococcal isolates. In 2019, Karna et al identified four E. faecium in five enterococcal isolates 9. Although E. faecium is a large-intestine symbiotic bacterium of humans and animals, it is listed by the World Health Organization as a glob-al priority for multidrug-resistant pathogens 10. In the present study, E. faecium isolates were resistant to vancomycin, erythromycin, clindamycin, ceftriaxone, and cefoxitin. Based on the published studies, 70% of isolated E. faecium strains from Tehran hospitals were resistant to vancomycin and erythromycin 11. In a similar study by Rahbar et al on clinical samples in Tehran, antibiotic resistance patterns of enterococcal isolates included 79% resistance to erythromycin and 51% to vancomycin 12. In this study, the prophage was identified in the E. faecium genome as reported in other studies 13.
In the present study, two recombinase family protein genes were reported. This protein catalyzes sensi-tive DNA exchange reactions between short target sequences (30-40 nucleotides). In 2010, Lopes et al reported the gene in the phage genome 14. Other genes identified in this study were hypothetical protein genes. The exact functions of these genes are unknown. In total, 25 hypothetical protein genes were reported in Entfac.YE prophage. Similar genes were identified in the EFRM31 phage genome by Mazaheri et al 15. Another gene was the tail tape measure protein, which determines the tail length and facilitates DNA transfer to the cell cytoplasm during cell infection. In the current study, two copies of this gene were identified. This gene was reported in 2016 in phage TP901-1 as well 16. In the present study, two copies of the tail protein gene were reported. This gene encodes proteins linked to the phage head. Three copies of gp6-like head-tail connector protein gene were identified in Entfac.YE prophage, encoding proteins that connect the phage head and tail. This gene was also identified in phages of Staphylococcus aureus (S. aureus) SA12 17. Two copies of the major capsid protein gene were identified in Entfac.YE prophage. As the gene name imparts, this gene encodes a capsid protein. In 2010, this gene was identified in the EFRM31 phage genome 15. In the present study, HK97 family phage prohead protease gene was reported. The major functions of this gene were breaking down scaffold proteins and processing capsid proteins. The mechanism of action of this gene was investigated in 2013 by Duda et al 18. Four copies of the portal protein gene were reported in the present study. The portal protein formed a channel for viral DNA to pass bilaterally. Another gene, the terminase large subunit gene, is involved in DNA transfer and packaging termination. Four copies of this gene were identified in Entfac.YE prophage. These genes were also reported in the EFRM31 phage genome 15. A similar gene, P27 family phage terminase small subunit gene, is responsible for binding to several identifying elements at the start of packaging. This gene has also been identified in S. aureus phages 19. A copy of HNH endonuclease gene was reported in the present study. The possible biological role of this gene is stimulating homologous recombination by nicking DNA, which enhances gene conversion. This gene was also reported in a study by Mazaheri et al 15. Two copies of MazG-like family protein gene were identified in Entfac.YE prophage which are involved in regulating the survival of bacterial cells under stress conditions. The function and structure of this gene in Deinococcus radiodurans were investigated by Goncalves et al 20.
A copy of helix-turn-helix domain-containing protein gene was identified in the present study. This protein can bind to DNA. In this study, two copies of antirepressor gene were reported. This gene prevents suppressor proteins from binding to their operators. This gene was identified in Salmonella phages as well 21. Nine copies of the site-specific integrase gene were reported in Entfac.YE prophage to rearrange DNA fragments. This gene was identified in the phage genome of TP901-1 22. A copy of YhgE/Pip domain-containing gene was identified in the phage genome. In the present study, a copy of helix-turn-helix transcriptional regulator gene, involved in transcription regulation, was characterized. Another gene, the recombinase RecT gene is involved in DNA binding and metabolic processing. A copy of this gene was reported in the phage genome. Functions of this gene in the phage genome of Bacillus subtilis were investigated in previous studies 23. A copy of ImmA/IrrE family metallo-endopeptidase gene was detected in Entfac.YE prophage. The ImmA protein, also encoded by transposons, has been shown to be essential for the breakdown of ImmR. The AAA family ATPase genes, identified in the present study, have a variety of roles such as cell cycle regulation, proteolysis and protein breakdown, and intracellular transport. The mechanisms of action of these genes were also investigated before 24. The function of the primase P4 family domain-containing gene includes nucleotide binding. The molecular function of the VRR-NUC gene includes domain-containing hydrolase activity that affects ester bonds. Based on previous studies, the exact function of this gene in phages is unknown 25. The tyrosine-type recombinase/integrase gene is involved in DNA binding and recombination, of which four copies were identified in the present study.
A domain version of the glucosaminidase domain-containing protein was reported in this study, which is responsible for the structural determination of peptides and glycans during vegetative growth. A copy of the phi13 family phage major tail gene was identified in Entfac.YE prophage and its molecular function was also investigated before 26. Two copies of ClpP protease gene were detected in the present study. This gene includes ATP-dependent peptidase and serine endopeptidase activities 27. Naturally, the ribbon-helix-helix domain-containing protein gene is involved in transcriptional regulation, a copy of which was identified in this study. Three copies of holin genes were identified in Entfac.YE prophage. Holins are a diverse group of small proteins produced by dsDNA phages to stimulate and control the destruction of the host cell walls at the end of the lytic cycle. Holins have been suggested as the clock proteins of phage infections 28. Two copies of the hemolysin XhlA family protein gene were identified in this study. XhlA is a cell surface-associated hemolysin that breaks down two common types of insect immune cells (Granulocytes and plasmatocytes) as well as rabbit and horse red blood cells. A copy of XkdX family protein gene reported in this study is detected in phage genomes close to choline and endolysin genes. The BppU family phage baseplate upper gene detected in the present study has been identified in the S. aureus phage genome. The virulence-associated protein E gene has also been identified in Streptococcus spp. 29. In the current study, a copy of the baseplate upper protein gene was reported. Based on the complete genomic analysis of an enterococcal phage by Mazaheri et al, genes similar to those of the present study were found, including genes of hypothetical protein, HNH endonuclease, tape measure protein, terminase small subunit, portal protein, prohead protease, major capsid protein, and major tail protein 15. In a study by Tan et al in 2007, genes similar to those of this study were reported, including terminase large subunit and portal protein genes 30. In the present study, 25 genes of hypothetical proteins were analyzed. In a study by O'Flaherty et al on the phage genome, 63 hypothetical protein genes were identified. Furthermore, they identified one AAA family ATPase, one endonuclease and one P27 family gene as well as one holin and one major capsid protein gene 31.
Conclusion :
Bacteriophages are naturally prevalent in the environment, especially in wastewaters. Nowadays, clinical bacterial isolates generally show resistance to antibiotics, which has created many problems for health professionals and patients. Prophages can be considered as mobile genetic elements in the transfer of antibiotic resistance genes to genomes of their host bacteria. Furthermore, genome analysis of prophages can help researchers better understand their roles in bacterial ecology and evolution.
Acknowledgement :
The authors thank all the staff within the Microbiology Laboratory, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran for their help.
Conflict of Interest :
The authors declare no conflict of interest.
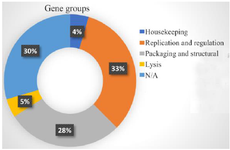
Figure 1. Analysis of the prophage gene groups.
|

Table 1. Information of the Illumina Hiseq platform
|

Table 2. An overview of the genes from Entfac.YE prophage
|

Count. Table 2. An overview of the genes from Entfac.YE prophage
CDS: Coding sequence; Gg: Gene group; bp: base pair; DDBJ: DNA Data Bank of Japan; NCBI: NCBI reference sequence; Cv: Coverage; Id: Iidentity; N/A: Not applicable; L: Lysis; RR: Replication and regulation; PS: Packaging and structural; H: Housekeeping.
|
|