Identification of a Novel Homozygous Mutation in BBS10 Gene in an Iranian Family with Bardet-Biedl Syndrome
-
Dehani, Mohammad
-
Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran
-
 Zare-Abdollahi, Davood
Fetal Health Research Center, Hope Generation Foundation, Tehran, Iran, Tel: +98 9138502360; E-mail: davood.zareabdollahi@gmail.com
Zare-Abdollahi, Davood
Fetal Health Research Center, Hope Generation Foundation, Tehran, Iran, Tel: +98 9138502360; E-mail: davood.zareabdollahi@gmail.com
-
Bushehri , Ata
-
Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran
-
Dehghani , Azadeh
-
Medical Biotechnology Research Center, Ashkezar Branch, Islamic Azad University, Ashkezar, Iran
-
Effati, Jalil
-
Meybod Genetic Research Center, State Welfare Organization, Yazd, Iran
Abstract: Background: Bardet–Biedl Syndrome (BBS) is a rare pleiotropic autosomal recessive disease related to ciliopathies with approximately 25 causative genes. BBS is a multisystemic disorder with wide spectrum of manifestations including truncal obesity, retinal dystrophy, male hypogenitalism, postaxial polydactyly, learning difficulties, and renal abnormalities.
Methods: A consanguineous Iranian family with a 28-year-old daughter affected with BBS, resulting from a first cousin marriage, was examined. After clinical examination, Whole Exome Sequencing (WES) was applied. Following the analysis of exome data, Sanger sequencing was used to confirm as well as to co-segregate the candidate variant with the phenotype.
Results: A novel homozygous variant [c. 2035G>A (p.E679K)] in exon 2 of the BBS10 gene was found which was categorized as likely pathogenic based on American College of Medical Genetics and Genomics (ACMG) guidelines and criteria. In this study, the variant was fully co-segregated with the phenotype in the family.
Conclusion: Despite overlapping with other ciliopathies in terms of the phenotype, the BBS has high genetic heterogeneity and clinical variability even among affected members of a family. The symptoms observed in patients are largely related to the genes involved and the type of mutations in the BBS. In this study, in addition to phenotype description of the proband harboring a novel disease-causing variant in BBS10 gene, the spectrum of BBS symptoms was expanded. The findings of this study can be useful in genetic counseling, especially for risk estimation and prenatal diagnosis.
Introduction :
Bardet–Biedl Syndrome (BBS; OMIM# 209900) is a rare pleiotropic autosomal recessive disease characterized by learning difficulties, male hypogenitalism, renal abnormalities, retinal dystrophy, obesity, and postaxial polydactyly 1. Secondary features may include metabolic and cardiovascular defects, hearing loss, ataxia, anosmia, diabetes, speech deficits, strabismus, hypertension, and dental malformations 2,3. At present, approximately 25 causative genes have been identified that can explain molecular causes in about 80% of affected families 4-6. Among these BBS-associated genes, the most involved genes are BBS1 (23.2%), BBS10 (20%), BBS2 (8.1%), BBS9 (6%), MKKS (5.8%), BBS12 (5%), and MKS1 (4.5%) 7-10. BBS is a rare disorder with a prevalence of about 1 in 150000 individuals worldwide 11. The criteria needed to clinically confirm the disorder upon examination include three or four main in addition to at least two minor symptoms of the BBS 12,13. In most patients, the inheritance pattern of BBS is autosomal recessive but nearly 10% of the BBS subjects have digenic triallelic inheritance 14. BBS belongs to a class of ciliopathies that are the biogenesis and the function of cilia 15. BBS10 gene has two exons and a length of 23.97 Kb 2. BBS10 protein consists of 723 amino acids and has three functional domains: equatorial, intermediate, and apical 3. The purpose of this study was to report a 28-year-old woman with BBS with a novel homozygous mutation [c.2035G>A (p.E679K)] in the second exon of BBS10 gene.
Materials and Methods :
Patients: A consanguineous Iranian family with symptoms of BBS disease was enrolled in this study. The family was from Yazd province and had only one affected 28-year-old daughter and 5 unaffected children (Figure 1). Clinical examinations were carried out by a clinical geneticist that verified three main features of learning problems, retinal dystrophy (Retinitis pigmentosa), and obesity in the patient (Figure 2A and 2B). Moreover, there were other manifestations in the patient such as hypothyroidism, hypertension, seizure, developmental delay, mild ID, and lingual problem. Additionally, for confirmation of the retinitis pigmentosa phenotype, ophthalmic examinations were performed including Optical Coherence Tomography (OCT) and fundus photography (Figure 2C-E). Informed consent was obtained from the patient and her family. This study was consistent with the Helsinki Declaration and methods accepted by the Research Ethics Committee (REC) (Approval number 1396.333) of University of Social Welfare and Rehabilitation Sciences (USWR) 16.
Mutation analysis: At first, 6 ml of blood sample from peripheral blood lymphocytes was taken from the patient, her parents, and other participants of the family and then genomic DNA was extracted by salting out method. Quality of the extracted DNA was examined by NanoDrop machine. Next, the sample was sent for Whole Exome Sequencing (WES), based on Next Generation Sequencing (NGS), to determine the sequence. Agilent SureSelect Human All Exon V6 (Agilent Technologies Inc., USA) was employed for exon enrichment. For sequencing reads, Illumina HiSeq 4000 platform (Seoul, Korea) was used at mean depth of coverage of 80X. The Burrows-Wheeler Aligner was applied for aligning the sequence reads with GRCh38 reference genome (https://www.ncbi.nlm.nih.gov/grc/human data). Furthermore, the processing of variant calls was completed by the Genomic Analysis Tool Kit (GATK). In data analysis, mutations based on exonic, exonic splice, splicing, frameshift, nonsynonymous, stop gain and stop loss variants with a nucleotide conservation score of GERP++>+2 and CADD>20 were selected and then according to the inheritance pattern compatible with autosomal recessive mode, the heterozygous variants were removed 17. In the next step, variants with a frequency of more than 0.005% were excluded based on the databases including HEX (https://www. alzforum.org/exomes/hex), EVS (http://evs.gs.wash-ington.edu/EVS/), 1000 genomes (http://www.interna-tionalgenome.org/), gnomAD (http://gnomad.broadin-stitute.org/), ExAC (http://exac.broadinstitute.org/), and Iranian national genome database (Iranome; http:// iranome.ir/). After filtering the variants, a novel homozygous mutation (c.2035G>A (p.E679K)) was found in exon 2 of the BBS10 gene. This variation is considered as a likely pathogenic mutation based on the American College of Medical Genetics and Genomics (ACMG) guidelines and criteria 18. The pathogenicity of c.2035G>A mutation was examined by some in silico tools like SIFT 19, polyphen-2 20 and Mutation Taster 21. For confirmation of the segregation and candidate mutation in all the family members, Primer3 (http:// frodo.wi.mit.edu/primer3/) was applied to design the primer set for second exon of BBS10 gene including forward primer 5'-GCTGGTTGTGTTTTGCCAGT-3', and reverse primer 5'- ATGAAGGAGGGCTGGAGTGA. After amplification of BBS10 gene using the polymerase chain reaction, Sanger sequencing was applied in order to sequence the amplicons using ABI BigDye terminator and ABI 3730×l DNA Analyzer (Applied Biosystems, USA). In this study, CodonCode Aligner 8.0.2 was used to analyze the sequencing results.
Results :
As mentioned, one BBS patient from a consanguineous marriage was examined. WES was performed on the patient and data analysis led to the identification of a novel putative homozygous mutation [c. 2035G>A (p.E679K)] in BBS10 gene. This mutation based on ACMG guidelines is considered as a likely pathogenic mutation. Also, further analysis by various in silico tools demonstrated that this change could be considered as a result of a disease-causing variant. Furthermore, the CADD and GERP ++ scores indicate the high conservation of this locus during evolution. Therefore, missense mutation at this locus could potentially have a detrimental effect on protein function and eventually lead to pathogenicity (Table 1). Then, PCR products sequenced by Sanger sequencing confirmed the presence of this mutation. Additionally, segregation analysis revealed that both parents were heterozygous for the mutation transmitted from them to the patient, which is indicated in figure 1B.
Discussion :
BBS is a rare genetic disorder with 25 genes identified in this disease 5,6. In this study, c.2035G>A mutation was detected in the BBS10 gene. Notwithstanding the fact that variable mutations occur in BBS10, the most common mutation in this gene is p.C91LfsX5 12. BBS1 and BBS10 genes have a high incidence of mutation worldwide whereas the studies conducted in Iran showed that BBS4 and BBS7 genes have the greatest role in occurrence of the disease in Iranian population 7. Clinical examinations on the patient revealed that she had three out of the six main symptoms, along with several other secondary symptoms. Among the primary features, the patient had truncal obesity, learning difficulties, and retinal dystrophy. Although the sign of postaxial polydactyly is found in most BBS patients, our patient did not exhibit the symptom (Figure 2F) 22. The detailed ophthalmologic examination, along with OCT and fundus photography indicated that the vision problem was actually retinitis pigmentosa, followed by night blindness, visual field defect, progressive photophobia, photopsia, color vision defect, and impaired visual acuity. The patient exhibited secondary complications including hypertension, developmental delay, and lingual problem. Additionally, in our patient, few other symptoms were displayed such as tonic-clonic seizure as well as hypothyroidism, which have not been mentioned in the literature, suggesting that c.2035G>A mutation in the BBS10 gene could lead to such symptoms.
Conclusion :
In our study, after identification of a novel homozygous variant c.2035G>A in BBS10 gene, two phenotypes of seizure and hypothyroidism were found that were not reported in previous studies. Due to scarcity of data and also high genetic and clinical heterogeneity of this disease, these two phenotypes can be regarded as symptoms of BBS. Given that few studies have been carried out so far on BBS patients in Iran, further studies should be implemented to confirm the above findings and understand different aspects of the disease besides its symptoms in Iranian community in order to improve prenatal diagnosis and apply the findings in genetic counseling.
Acknowledgement :
We would like to acknowledge the participation of the daughter, her family, and the colleagues who helped us in this study.
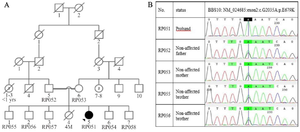
Figure 1. Pedigree and sequencing data of the family. A) This pedigree displays a BBS patient from a consanguineous marriage who is the only one affected in her family, B) As shown in chromatograms, proband was homozygous for c.2035G>A mutation and inherited mutation from both carrier parents. Also, two normal brothers (RP055 and RP056) who participated in this study were carriers of mutation.
|
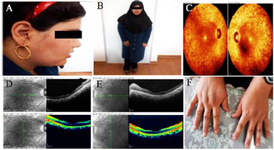
Figure 2. Clinical symptoms of BBS proband. Funduscopy (A) and OCT (B and C) of both right and the left eyes of the affected case. Clinical features of proband (D and E) without polydactyly (F).
|

Table 1. In silico analysis of c.2035G>A (p.E679K) mutation in BBS10 gene
MAF: Minor allele frequency; N/A: Not available
|
|