Construction and Evaluation of Short Hairpin RNAs for Knockdown of Metadherin mRNA
-
Zare, Farahnaz
-
Division of Medical Biotechnology, Department of Medical Laboratory Sciences, Faculty of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
-
Diagnostic Laboratory Sciences and Technology Research Center, Faculty of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
-
 Sharifzadeh , Sedigheh
Division of Medical Biotechnology, Department of Medical Laboratory Sciences, School of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran, and Diagnostic Laboratory Sciences and Technology Research Center, Faculty of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran, Tel: +98 71 32270301; E-mail: sharifsd@sums.ac.ir
Sharifzadeh , Sedigheh
Division of Medical Biotechnology, Department of Medical Laboratory Sciences, School of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran, and Diagnostic Laboratory Sciences and Technology Research Center, Faculty of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran, Tel: +98 71 32270301; E-mail: sharifsd@sums.ac.ir
-
Behzad-Behbahani, Abbas
-
Division of Medical Biotechnology, Department of Medical Laboratory Sciences, Faculty of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
-
Diagnostic Laboratory Sciences and Technology Research Center, Faculty of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
-
Rafiei Dehbidi, Gholamreza
-
Diagnostic Laboratory Sciences and Technology Research Center, Faculty of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
-
Yousefi, Zahra
-
School of Allied Medical Sciences, Shahroud University of Medical Sciences, Shahroud, Iran
-
Ranjbaran, Reza
-
Diagnostic Laboratory Sciences and Technology Research Center, Faculty of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
-
Seyyedi, Noorossadat
-
Division of Medical Biotechnology, Department of Medical Laboratory Sciences, Faculty of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
-
Diagnostic Laboratory Sciences and Technology Research Center, Faculty of Paramedical Sciences, Shiraz University of Medical Sciences, Shiraz, Iran
Abstract: Background: Short hairpin RNA (shRNA) has proven to be a powerful tool to study genes’ function through RNA interference mechanism. Three different methods have been used in previous studies to produce shRNA expression vectors including oligonucleotide-based cloning, polymerase chain reaction (PCR)-based cloning, and primer extension PCR approaches. The aim of this study was designing a reliable and simple method according to the primer extension strategy for constructing four shRNA vectors in order to target different regions of Metadherin (MTDH) mRNA in human leukemic cell line Jurkat.
Methods: Oligonucleotides for construction of four shRNA vectors were designed, synthesized and fused to U6 promoter. Each U6-shRNA cassette was cloned into a pGFP-V-RS vector. MTDH shRNAs were transfected into the Jurkat cell line by using the electroporation method. The ability of shRNAs to knock down MTDH mRNA was analyzed through qRT-PCR. Apoptosis assay was used to evaluate the effect of down regulation of MTDH expression on cell integrity.
Results: A significant reduction (about 80%) in the expression levels of MTDH mRNA and an increase in the percentages of apoptotic cells (about 20%) were observed in the test group in comparison with control.
Conclusion: MTDH shRNA constructs effectively inhibited gene expression. However, simplicity and inexpensiveness of the method were additional advantages for its application.
Introduction :
Targeting mRNA molecules through RNA interference (RNAi) technology by double-stranded RNAs is a conserved mechanism to regulate and control gene expression 1,2. The first discovery of RNAi mechanism was in transfected cells from nematode Caenorhabditis elegans with double-stranded RNAs that could specifically target a gene of interest and potentially reduce its expression level 3.
Naturally, in the RNAi pathway, pri-miRNA is produced through cleavage of a long double-stranded RNA in nuclear by Drosha-DGCR8 (DiGeorge syndrome critical region 8) complex. Afterward, pri-miRNA is processed to mature short double-stranded RNA (21-22 nucleotides) by using the RNaseIII protein Dicer 4. This mature cleaved RNA (siRNA) is incorporated into RNA-Induced Silencing Complex (RISC) and then split into passenger and guide strands. The passenger strand is degraded and released from the complex. Guide strand leads the RISC to the specific sequence of the desired mRNA. The targeted mRNA is cleaved and degraded by nucleases and as a consequence, the target gene expression is downregulated or blocked 5.
Also, siRNA can be artificially synthesized in the form of double-stranded RNA that can be used to downregulate overexpressed genes in many diseases such as cancers and genetic disorders 6. Although siRNA has been introduced as a powerful product in the pharmaceutical industry, some biological barriers negatively influence its action. siRNA has a strong poly-anionic charge that directly inhibits its penetration into cells, and instead uses endocytosis or pinocytosis. Released siRNAs from endosomes undergo degradation by cytoplasmic ribonucleases. Furthermore, serum endonucleases degrade siRNAs after administration. Also, siRNAs have some non-target effects such as activation of innate immune systems and disturbing regulatory pathways in the cells through the displacement of miRNA from RISC. The other limitation of the siRNA application is that as cells divide, the concentration of siRNA decreases and becomes diluted 7. One of the common ways for producing siRNA is using vectors for expressing siRNAs as short hairpin RNAs (shRNA) under polymerase III promoters. U6 and H1 promoters are mostly used to express shRNA cassettes with 21-23 nucleotide sequence (Sense) similar to the target part of specific mRNA followed by a loop and 21-23 nucleotide complementary sequence (Anti-sense) 8,9. In contrast to siRNAs, shRNAs can be used for production of cell lines which stably knock down desired gene expression. To overcome the barriers of shRNA delivery from the membrane of almost all cell lines and to increase transfection efficiency, viral systems such as lentiviral-and adenoviral-based technologies can be used 10.
Several studies reported various methods for the construction of shRNAs expression vectors. Most shRNA vectors are constructed by synthesis, annealing, and ligation of two long oligonucleotides that contain hairpin sequence plus terminator 11. Synthesis of these long sequences increases the chance of mutation and also the cost of its application is higher than the other methods. In other methods, PCR based approaches, the promoter region is used as a template and amplified with one constant forward primer and different reverse primers. In this approach, the reverse primer contains a hairpin sequence that after amplification contains both promoter and hairpin cassettes. Production of this long oligonucleotide needs costly purification methods and also synthesizing sequences with secondary structures like hairpin increases the chance for mutations. To overcome these problems, two short reverse primers containing the sense and anti-sense sequences that are linked by loop sequence can be used and then by performing two consecutive PCR reactions, the resulting products will have promoter and hairpin sequences 12,13.
Metadherin (MTDH) is an oncogene and its increased expression is reported in cancerous cells from numerous tissues 14-16. MTDH is a 64 KDa transmembrane protein (Type II) expressed in the nucleus in normal cells, whereas in malignant cells, it is translated into the cytoplasm. MTDH protein can regulate some biological features of tumorigenic cells, such as cell survival, angiogenesis, metastasis, chemoresistance, and inhibition of cell apoptosis by modulating some cell signaling pathways such as PI3K/AKT, Wnt/β catenin, MAPK, and autophagy pathways 17-20. A recent study showed that MTDH is highly expressed in Acute Lymphocytic Leukemia (ALL) 21. In this study, a simple and cost-effective strategy was designed for constructing shRNA vectors to target MTDH mRNA in the T-ALL cell line (Jurkat cell line).
Materials and Methods :
Design and construction of shDNA cassettes: For efficient degradation of MTDH mRNAs, four shRNA expression cassettes (shDNAs) were designed to target different regions of the mRNA sequence. Additionally, for constructing each shDNA fragment, two oligonucleotides were synthesized that each of them contained 5´ -the last 12 nucleotides of U6 promoter-sense-loop-3´ and 5´-terminator (6 Adenine base)-sense-complementary loop-3´sequences (Table 1). They were permitted to anneal to each other via complementary regions in their 3´ ends. The ligation program included 10 cycles of 95°C for 30 s, 40°C for 30 s, and 72°C for 60 s. The constructs were amplified using sh.forward and sh.reverse primers (Figure 1A and Table 2), and PCR program involved 32 cycles at 95°C for 30 s, 58°C for 40 s, 72°C for 60 s.
Cloning shDNA cassettes into pGPF-V-RS vector: U6 promoter (247 bp length) was amplified from genomic DNA by using the U6.forward and U6.reverse primers (Table 2). In the next step, each of four shDNA fragments was attached to U6 promoter separately by overlap extension polymerase chain reaction (SOEing PCR). Briefly, in the first step (Primer independent annealing), two fragments (shDNA and U6 promoter) were permitted to be annealed by their complementary regions. In the second step (Primer dependent annealing), 0.5 µM from each U6 forward and sh.reverse primers were added to the PCR reaction. The PCR programs included 10 cycles of 95°C for 30 s, 40°C for 30 s, and 72°C for 60 s at step one, and 22 cycles of
95°C for 30 s, 60°C for 40 s, and 72°C for 70 s at step two. To reduce the rate of point mutations, all PCR reactions were performed using the Pfu DNA polymerase. The expected product was the U6-shDNA fragment that was digested with EcoRI and HindIII restriction enzymes (Jena Bioscience, Germany) and ligated into the pEGFP-V-RS vector (OriGene, USA) by using the T4 DNA ligase enzyme (Thermo Fisher Scientific, USA).
Cell culture: The human acute lymphoblastic leukemia cell line Jurkat was cultured in high glucose RPMI-1640 medium supplemented with 10% FBS (Gibco, USA) and 1% penicillin/streptomycin (Bioidea, Iran). Cells were maintained in humidified incubator with 5% CO2 at 37°C.
Transfection of constructed MTDH shRNAs into Jurkat cell line: Jurkat cells at passage three were used for transfection by using the electroporation method. Electroporation of cells was performed according to the following approach. The cells were harvested, centrifuged at 252×g for 5 min and washed three times with Phosphate Buffered Saline (1×PBS) (Sigma Aldrich, USA). About 4×106 cells were loaded into the Bio-Rad 2 mm electroporation cuvette, mixed with 12 µg MTDH shRNA vectors and pulsed (Voltage of 280 V and electric capacity of 600 µF) using Gene Pulser Xcell™ Electroporation Systems (Bio-Rad, USA). All four constructed shRNAs were entered into the electroporation cuvette simultaneously. Next, cells were resuspended in complete media, transferred into a 25 ml tissue culture flask and incubated for 72 hr for further analysis. Transfection efficiency was determined by means of fluorescence microscopy. The transfected cells with scrambled shRNA plasmids were used as the control group. The experiments were performed independently in triplicate manner.
RNA extraction and cDNA synthesis: To evaluate the efficiency of shRNAs in reducing the MTDH gene expression, 72 hr after transfection of Jurkat cells with MTDH shRNAs, cells were harvested and total RNA was extracted using RNXplus solution (CinnaGen, Iran) according to the manufacturer's recommendations. The concentration (ng/µl) and purity (Absorbance at 260 nm/280 nm) were measured by spectrophotometer and nanodrop cuvette (Hellma, Germany).
cDNA synthesis was performed using Takara kit (Takara, Japan) according to the manufacturer's guide-lines. Based on the concentration of extracted total RNA, 1000 nanograms of RNA were used in the cDNA synthesis reaction.
Real-time PCR: Firstly, specific primers were designed using Gene Runner software. Then, amplification of target genes was performed using SYBR Green PCR Master Mix (SYBR Premix Ex Taq™II, Tli RNaseH Plus, Yekta Tajhiz, Iran) and specific primers (Table 3) on real-time PCR cycler, the Rotor-Gene Q (Qiagen, USA). The β-actin was served as a housekeeping gene and used for the normalization of MTDH gene expression. The qRT-PCR program included first denaturation step at 95°C for 90 s followed by 45 cycles of denaturation at 95°C for 30 s, annealing at 60°C for 30 s and elongation steps at 72°C for 30 s.
Flow cytometry analysis of apoptosis: The effect of shRNA constructs on apoptosis of Jurkat cells was assessed using the PE-Annexin V/7AAD kit (BD Biosciences, USA). Briefly, cells were harvested, washed with 1×PBS, and resuspended in 1X binding buffer. Cells were incubated with 5µl of 7AAD and 2.5 µl of Annexin V for 15 min on ice and in dark. Then, 400 µl of binding buffer was added to each tube and the percentage of apoptosis was assessed using BD FACSCalibur flow cytometer (BD Biosciences, USA) and data was analyzed by FlowJo software v7.6.
Statistical analysis: Data analysis was performed using the GraphPad prism v7.03 software. Analysis of real-time PCR results was conducted using one-sample t-test. A comparison of apoptosis in MTDH shRNA-transfected cells and scrambled shRNA-transfected cells was performed by using the two-tailed unpaired t-tests. The p-values less than 0.05 were considered statistically significant.
Results :
Design and construction of shRNA expression vectors: The purpose of the study was to design a reliable, cost-effective, and trouble free method for producing shRNA expression vectors. Among the different available strategies, primer extension PCR was selected. Four shRNA vectors were designed for targeting different regions of MTDH mRNA. Two short oligonucleotides with the complementary region at their 3' ends were synthesized for each shRNA and permitted to generate double-stranded fragments with 5´-sense-loop-antisense-3´ sequence. Afterward, these fragments were amplified and electrophoresed using 1.5% agarose gel. The presence of a 105 bp band in agarose gel following ethidium bromide staining confirmed the generation of the desired product. In the next step, the U6 promoter sequence was amplified with specific primers from genomic DNA and fused to shDNA cassettes. U6-shDNA fragments were inserted into the pGFP-V-RS vector and colony PCR was performed to evaluate the accuracy of positive clones (Figure 1B).
Efficient silencing of MTDH mRNAs: To determine the efficiency of constructed shRNA vectors for knockdown of MTDH mRNAs, Jurkat cells were transfected with these constructs through the electroporation method (Figure 2). The qRT-PCR assay was performed to evaluate the downregulation of MTDH mRNAs in 72 hr post-transfection. The results revealed that expression levels of MTDH mRNA significantly decreased about 80% after the treatment of cells with MTDH shRNAs in comparison to the control group (p<0.05) (Figure 3).
Apoptosis assay: For assessment of the MTDH shRNAs ability to induce cell death, the apoptotic status of cells was analyzed using PE-Annexin V/7AAD reagents. Flow cytometry analysis showed that apoptosis in Jurkat cells increased (about 20%) in transfected cells with MTDH shRNAs compared with the control group (50.04± 2.884, mean±SD vs. 19.13±6.586, mean±SD; p= 0.0127) (Figure 4).
Discussion :
RNAi strategy is a natural tool that can be used for targeting oncogenes in tumorigenic cells. RNAi is mediated by double-stranded small RNAs such as miRNAs, siRNAs, and shRNAs 22. Unlike siRNAs, shRNAs have long-term effects with low turnover because of continuous production in the host cell. On the other hand, a low copy number of shRNAs is required for efficient gene silencing that contributes to decreased off-target effects in comparison with siRNAs 23.
Preparation of shRNA expression vectors can be done according to different methods, including oligonucleotide-based cloning and PCR-based cloning methods 10,24. In many studies, long synthetic oligonucleotides which contain hairpin sequence are used. These oligonucleotides are allowed to be annealed through their complementary region and then cloned into desired vectors. Though this is a fast strategy, the cost of synthesizing long sequences is higher than other methods, and also the possibility of mutation within the hairpin region increases about 20-40% 11. In other methods, PCR based strategies, a promoter serves as a template, and amplification takes place by using a long reverse primer which contains a hairpin sequence. Although this method is performed by using one round of PCR reaction, synthesizing a long oligonucleotide with hairpin structure is hard and susceptible to mutations. Also, this long sequence needs costly purification methods such as the PAGE method to eliminate damaged and truncated sequences 12,13. In the current study, an efficient and simplified method was developed to generate shRNA expression vectors by using the two short oligonucleotides. In the first step, two short oligonucleotides were designed that contained 5´-the last 12nt of U6 promoter, sense (29 nt) and loop (9 nt) -3´, and 5´-sense (29 nt)-complementary loop (9 nt) and terminator (6 nt) sequences. These oligonucleotides annealed through their complementary region at their 3´ ends and DNA polymerase extended them to construct a double-stranded DNA with complete hairpin sequence (Sense, loop, anti-sense, and terminator). Next, the U6 promoter was amplified from genomic DNA and fused to upstream of shDNA fragments by performing SOEing PCR reaction and cloned into the pGFP-V-RS vector. For reducing the mutation rate, Pfu DNA polymerase was utilized in all PCR reactions.
Due to the formation of secondary structures, it is difficult to confirm the accuracy of sequences of constructed shRNA vectors. Some studies show that secondary structures like hairpin structure can cause premature termination of sequencing reaction by disrupting the processing of DNA polymerase during sequencing reaction. Many researchers tried to improve sequencing conditions through different approaches 25,26. Due to a lack of access to the sequencing instrument, accuracy of shRNA sequences could not be directly evaluated in this study. Therefore, to confirm the ability of shRNA vectors to down-regulate expression of a specific gene, MTDH mRNA was selected as a target gene to design and construct shRNA expression vectors. MTDH is an oncogene and the findings show its overexpression in cancerous cells of breast, colorectal, neuroblastoma cells, and blood and bone marrow cells 14,16. Hence, Li et al in 2020 reported that the expression of MTDH highly increased in primary T-cell Acute Lymphoblastic Leukaemia (T-ALL) and T-ALL cell line (Jurkat) 21. In our study, an attempt was made to evaluate the capability of constructed shRNA vectors for silencing MTDH mRNAs by transfecting them at the same molarity into Jurkat cells using the electroporation method. Real-time PCR results demonstrated that MTDH mRNA expression decreased by about 80% in transfected cells with constructed MTDH shRNA vectors in comparison with control cells. Besides, flow cytometry analysis showed that apoptosis of Jurkat cells increased about 20% after treatment of cells with MTDH shRNAs compared to the control group which was transfected with scrambled shRNA plasmids. As expected, these results indicate that shRNA constructs were accurately synthesized.
Conclusion :
In conclusion, these findings demonstrated a simple, low cost, and fast method with effective silencing power for the construction of shRNA expression vectors. The main limitation of the study was setting up a sequencing reaction specifically for shRNA sequence analysis. It seems that further research is required to approve the potential of this method for generation of RNA interfering molecules.
Acknowledgement :
This manuscript was extracted from the MSc thesis of Farahnaz Zare. It was supported by a grant (No: 94-01-10-9281) from the Vice-Chancellor for Research Affairs of Shiraz University of Medical Sciences, Shiraz, Iran. We are also grateful to all staff of the Diagnostic Laboratory Sciences and Technology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran for their technical assistance.

Figure 1. Designing shDNA fragments, A: 1) The first oligonucleotide contains 3´ end of U6 promoter, sense and loop sequences, and the second oligonucleotide contains 6 Adenine nucleotide to generate terminator, sense, and complementary sequences for loop region sequences. These oligonucleotides were annealed at their 3´ ends and extended to make shDNA fragments, 2) The amplified U6 promoter was fused to shDNA via SOEing PCR. B: Gel electrophoresis of 1) Amplified shDNA fragments by using the sh.forward and sh.reverse primers (103 bp), 2) Amplified U6 promoter (247 bp), and 3) Amplified U6-shDNA fragments using sh.forward and U6.reverse primers (330 bp).
|
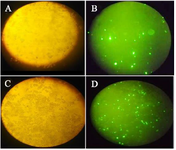
Figure 2. For evaluation of transfection efficiency, GFP expression was detected using fluorescence microscopy. A) Light and B) fluorescence microscopy images of transfected cells with MTDH shRNAs, C) Light and D) fluorescence microscopy images of control cells that were transfected with scrambled shRNA plasmid.
|
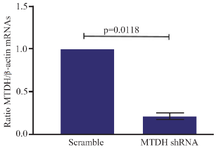
Figure 3. shRNA mediated downregulation of MTDH mRNA. The levels of MTDH mRNA, 72 hr after treatment of Jurkat cells with MTDH-shRNAs or treatment of the control group with scrambled shRNA plasmids. The Two-tailed one-sample t-test was used for data analysis. MTDH expression decreased after being targeted with constructed MTDH shRNAs. Expression values of MTDH mRNA are presented as mean±SD from three independent experiments (p= 0.0118).
|
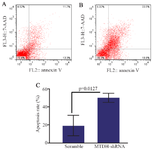
Figure 4. The effect of MTDH shRNAs on the apoptosis of Jurkat cells, A) PE-Annexin V/7AAD staining was performed for determining the effect of MTDH mRNA silencing on the apoptosis of Jurkat cells, B) Transfected cells with scrambled shRNA plasmids were used as a negative control, C) Data analysis of apoptosis assay. The values were analyzed using the two-tailed unpaired t-test and are presented as mean±SD from three independent experiments (p= 0.0127).
|

Table 1. Sh.oligonucleotide sequences. The underlined nucleotides are sense sequences for targeting MTDH RNA
|

Table 2. Primer sequences for amplification of shDNA cassettes. The underlined nucleotides are HindIII restriction sites
|

Table 3. Primer pairs used for quantitative real-time PCR
|
|