Benchmarking Datasets from Malaria Cytotoxic T-cell Epitopes Using Machine Learning Approach
-
 Adiga, Rama
Nitte (Deemed to be University), Nitte University Centre for Science Education & Research (NUCSER), Division of Bioin-formatics and Computational Genomics, Deralakatte, Paneer Campus, Mangalore, India 575018, Tel: +91 824 2204300 lines (1-4), E-mail: rama_adiga@nitte.edu.in
Adiga, Rama
Nitte (Deemed to be University), Nitte University Centre for Science Education & Research (NUCSER), Division of Bioin-formatics and Computational Genomics, Deralakatte, Paneer Campus, Mangalore, India 575018, Tel: +91 824 2204300 lines (1-4), E-mail: rama_adiga@nitte.edu.in
Abstract: Background: Epitope prediction remains a major challenge in malaria due to the unique parasite biology, in addition to rapidly evolving parasite sequence variation in Plasmodium species. Although several models for epitope prediction exist, they are not useful in Plasmodium specific epitope development. Hence, it was proposed to use machine learning based methods to develop a peptide sequence based epitope predictor specific for malaria.
Methods: Model datasets were developed and performance was tested using various machine learning algorithms. Machine learning classifiers were trained on epitope data using sequence features and comparison of amino acid physicochemical properties was done to yield a valid prediction model.
Results: The findings from the analysis reveal that the model developed using selected classifiers after preprocessing by Waikato Environment for Knowledge Analysis (WEKA) performed better than other methods. The datasets for benchmarks of performance are deposited in the repository https://github.com/githubramaadiga/epito-pe_dataset.
Conclusion: The study is the first in-silico study on benchmarking Plasmodium cytotoxic T cell epitope datasets using machine learning approach. The peptide based predictors have been used for the first time to classify cytotoxic T cell epitopes in malaria. Algorithms has been evaluated using real datasets from malaria to obtain the model.
Introduction :
Increasing availability of data and the advancement of machine learning methods have resulted in improved performance of these methods in recent years, as has been demonstrated on Major Histocompatibility Complex (MHC) binding data 1-5. MHCs bind and present peptides to T cells for recognition. Many antigens with potential T cell epitope have been mapped in Plasmodium and reviewed 4. Several methods for in silico quantitative prediction of MHC class exist 6-11 which do not provide reliable prediction scores. Based on the prediction of MHC binding required for T cell recognition, specific epitopes have been reported. Though experimentally it is a very time consuming extensive process, bioinformatics methods may help to develop epitope predictors rapidly and accurately.
Various machine learning algorithms are available for testing model performance in predicting epitopes. Some commonly used algorithms in WEKA include Naive Bayes, k-nearest neighbour, logistic regression, and support vector machines (SVM). Naive Bayes is an implementation of Bayes’ theorem supporting multi-class and binary classification problems. The KNN algorithm and logistic regression are simple methods for generalization in small samples and in using binary classification 12. Sequential Minimal Optimization (SMO) refers to the optimization algorithm used within the SVM implementation. Naive Bayes and SVM are generally found to be suitable for bioassay based work 13 but do not handle class overlap very well. Supervised machine learning algorithms which are ensemble-based perform better than individual classifiers and are increasingly being used. Ensemble methods are the meta algorithms generating one predictive model and decreasing bias and variance, thereby improving prediction ability. In the current study, the use of meta classifiers was evaluated and discussed.
There has been a recent explosion of data driven solutions for the prediction of T-cell epitope. The peptide based sequence information has been exploited leading to various machine learning algorithms being tested and validated. However, in the area of malaria studies, research on peptide based sequence information is scarce. Thus, in this study, a benchmark training set was developed which would be unique and it provides a comparative metric for testing the performance and evaluation of algorithms. The present analysis was performed for cytotoxic T-cell epitopes of Plasmodium.
Materials and Methods :
Preparation of dataset: Dataset preparation requires a well-curated and ambiguous dataset which can be used for model building using machine learning. Data mining literature for epi-tope regions of Plasmodium species available in published literature was evaluated. Plasmodium epitopes were extracted and their allelic association was also collected from published literature with experimental validation 14-16. The datasets 1 and 2 comprised 54 and 16 cytotoxic T cell epitopes, respectively obtained from studies conducted by Wizel et al 17, Doolan et al 14 and Carralot et al 16. The literature showed the peptide sequence to be naturally associated with HLAA2, A3, B7, B8 or DRB1 allele. In the present study, dataset 1 was used as the main input data where 80% of the dataset was used for training and the remaining 20% for testing. Dataset 2 was used for validation of models. The datasets were subjected to feature selection and the dipeptide composition profile was created as described below. The details of preprocessing are also given below. The step by step guide on using WEKA and installing the library intended for machine learning are available at https://wekatutorial.com/# functionalityandfeatures.
Input features: Feature selection: Input features were generated using a widely used peptide feature, mainly dipeptide composition. Dipeptide composition accommodates more information due to longer vector length of 400 18,19. The present study highly relies on the feature for classification. The Pfeature (https://webs.iiitd.edu.in/ raghava/pfeature/) is a popular webserver for computation of features from peptide sequences. The average score was calculated from the 400 dipeptide composition features extracted from Pfeature. It provides the composition of each of the possible 400 dipeptides formed by 20 amino acids. The simple dipeptide composition profile was created for the dataset using 400 dipeptides for alleles. The methods are depicted in a workflow (Figure 1).
Preprocessing of dataset: The average dipeptide score was calculated for each HLA allele which was used for further processing. The profile for the input data was extracted from Pfeature for each of the peptide sequences of datasets 1 and 2. Average values were used to generate a new table. The minimum number of sequences used for dipeptide composition analysis was set to 6 sequences. Two sequences associated with B7 and B8 allele did not fulfil the criteria which were removed and not considered for further analysis. Another sequence from dataset 1 was not considered since the allelic information of the peptide was not available.
Attribute discrimination method: The stable version of WEKA 3.8 was downloaded and installed along with the associated packages 20. The reduction in number of attributes was possible by using the "select attributes" menu of WEKA.
Machine learning based prediction models: Cross-validation: A five fold cross-validation technique was employed for training, testing and evaluation. In summary, the technique involves dividing the dataset into five equal parts. Four sets would be used for training, while the remaining set for testing. After iterating five times, each set is used for testing.
Results and Discussion :
Compositional analysis of peptides was carried out in the study and it was observed that the dipeptide which was classified as HLA A2 in the dipeptide composition analysis had 63% hydrophobic residues (Table 1) and 13% acidic residues (Cutoff >1.4) whereas HLAA3 had only 63% hydrophobic residues (Cutoff >1.6). Similarly, dipeptides from DRB1 epitopes had 43% hydrophobic and 22% basic residues (Cutoff >1.8) while those classified as non-epitopic had only 37% hydrophobic residues and 16.6% acidic residues (Table 1). The difference in physicochemical properties suggested that the information can be used for classification.
Machine learning based classification: The analysis of dipeptide composition profile may be exploited for purpose of classification of malaria cytotoxic T cell epitopes into allele based sequence specific features where the local order of amino acids is preserved. Machine learning based classification was implemented to exploit the sequence specific features for the classification and prediction of malaria epito-pes.
Dipeptide composition based model: A Pfeature peptide based prediction was constructed to obtain the possible dipeptide combinations in a 400 sized vector. The average score was considered as input feature in WEKA. Individual prediction model was built from feature set and compared after normalization.
The large number of attributes (400) needed to be reduced and filtered to keep only the relevant attributes. For dimensional reduction, the WEKA CfsSubSetEvaluator Algorithm and the Ranker Search Method were used. The Correlation Attribute Evaluator was applied sequentially and the top 26 attributes were ranked and used for further analysis (Table 2).
Comparison of machine learning methods: Ensemble classifiers have been known to be advantageous and outperform single decision tree based classifiers by having higher accuracies and smaller prediction errors. In the current study, simple logistic regression or SVM based methods were not suitable as classifiers because of class overlap. Hence, it was decided to use meta classifiers.
The boosting algorithms are a way of combining many weak algorithms into an additive logistic regression. AdaBoost uses a majority vote in the weighted version of sequentially applied classifiers. The AdaBoost meta-estimator fits the classifier on the dataset adjusting weights for instances which are incorrectly classified. AdaBoost and Logitboost generally achieve comparable level of performance and result in high degree of accuracy.
AdaBoostM1: An ensemble technique of boosting has been used with a number of weak classifiers to create a strong prediction based classifier. Its similarity with Random Forest is that it uses decision trees for final classification within the forest. The decision trees in AdaBoostM1 have a depth of 2 leaves.
Iterative Classifier Optimizer: Another meta classifier uses neural network and compares the actual classification of the record and optimizes it. The algorithm is modified for further iterations by feedback error system obtained from the classification of the first record.
Iterative Classifier Optimizer Algorithm from meta classifier of WEKA: This algorithm chooses the number of iterations (Default of L=50) which was found to be suitable for classifying. Logitboost was selected as the default Iterative Classifier for cross-validation or a percentage split evaluation. A 10 fold cross-validation was performed for the required number of runs with default of R=1.
Model development: The training model was developed by preprocessing with unsupervised filter class and converting string attributes into numeric class. The StringToWordVector filter creates a separate dictionary and merges them. The file was saved and various classifiers were tested. The confusion matrix for the meta classifier performed better than others and was able to correctly classify 14 attributes (53.8%) (Table 3). The Area Under the ROC Curve (AUC) of the model for being classified as HLAA2, HLAA3, DRB1 or non-epitopic was 0.73, 0.80, 0.68 and 0.71, respectively. Additional class of HLAA2/HLAA3 indicated that few peptides were classified as both classes with AUC of 0.68 and some peptides as HLAA3/DRB1 with AUC of 0.80 (Table 4). The precision value was1.0 for meta classifier.
Performance of model: The model classifier performed an internal 10 fold cross-validation procedure (set to 10) which was inbuilt in the Iterative Classifier Optimizer algorithm of meta classifier. Hence, the model obtained was a 10 fold cross-validated model.
Validation for this model was done using data extracted from an independent study15 and annotated as dataset 2. Data validation for dataset 2 was done by extracting features and performing similar processing as described in methods. All the 36 instances were used for feature selection. It was subjected to cross-validation and 30 instances were correctly identified (83% accuracy).
Performance of model on independent dataset: The performance of model was evaluated on dataset obtained from independent study. Pre-processing was performed as mentioned above and the OneR classifier was used with "re-evaluate model on current test set" option of WEKA to obtain 100% accuracy, ignoring 6 instances in the classification of HLAA2 with ROC value of 0.500 and precision of 1.00. The accuracy for the model during performance evaluation was skewed toward classification of HLAA2 since it included those alleles in the dipeptide composition profile of the dataset.
Performance of model using SMOTE: Synthetic Minority Over-sampling TEchnique (SM-OTE) gives improved performance by oversampling without losing the data. The SMOTE filter was applied (400%) to increase the weight of classes and the number of attributes increased by 8 instances totalling 34. Out of this, 18 were correctly classified (52.9%). The AUC values obtained were 0.906 and 0.627 for classifying allele HLAA3 and DRB1, respectively. Other alleles could not be classified. Thus, SMOTE filter for oversampling was not suitable for the study and did not perform well.
Conclusion :
The primary sequence based information preserves the local order of molecules which is the superior method of evaluation. In the current study, Iterative Classifier Optimizer algorithm performed better than Logitboost or AdaBoostM1. Further studies will help in developing a model and will further improve identification of malaria epitope. The unique characteristics of the biology of the parasite and the sequence variation therein would make machine learning technique ideal for Artificial Intelligence (AI) based application for fast and rapid detection of epitopes.
Acknowledgement :
The author wishes to thank Dr. Anirban Chakra-borty, Director of Nitte University Centre for Science Education and Research (NUCSER), Prof. Dr. Indrani Karunasagar, Director (Projects) and the management of Nitte University, Deralakatte, Mangalore, Karnataka, India. This study was supported by Nitte University grant number NUFR2/2018/10/26.
Conflict of Interest :
There are no conflicts of interest reported.
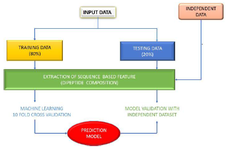
Figure 1. Methodology workflow used in the study.
|
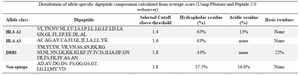
Table 1. Dipeptide composition profile generated from dataset 1
|
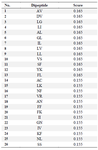
Table 2. Feature selection using WEKA CfsSubSetEvaluator Algorithm for selecting attributes and ranking by Correlation Attribute Evaluator and Ranker Search Method
|

Table 3. Confusion matrix generated using WEKA Iterative Classifier Optimizer Algorithm for 26 instances (14 were correctly classified) showing 6x6 confusion matrix to describe six classes assigned a to f (Sum of diagonals indicated the number of correctly classified instances)
|
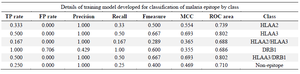
Table 4. Accuracy of training model by class using Iterative Classifier Optimizer
|
|