Using CRISPR/Cas9 System to Knock out Exon 48 in DMD Gene
-
Dara , Mahintaj
-
Department of Molecular Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
-
Razban, Vahid
-
Department of Molecular Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
-
Talebzadeh, Mahdieh
-
Department of Molecular Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
-
Moradi, Sepideh
-
Department of Molecular Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
-
 Dianatpour, Mehdi
Department of Medical Genetics, Shiraz University of Medical Sciences, Shiraz, Iran, Tel/Fax: +98 713 2349610, E-mail: dianatpour@sums.ac.ir
Dianatpour, Mehdi
Department of Medical Genetics, Shiraz University of Medical Sciences, Shiraz, Iran, Tel/Fax: +98 713 2349610, E-mail: dianatpour@sums.ac.ir
-
Department of Medical Genetics, Shiraz University of Medical Sciences, Shiraz, Iran
-
Stem Cell Technology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
Abstract: Background: Out of frame mutations in DMD gene cause Duchenne Muscular Dystrophy (DMD) which is a neuromuscular progressive genetic disorder. In DMD patients, lack of dystrophin causes progressive muscle degeneration, which results in heart and respiratory failure leading to premature death. At present, there is no certain treatment for DMD. DMD gene is the largest gene in human genome by 2.2 mega base pairs and contains 79 exons. In the past few years, gene therapy has been considered a promising DMD treatment, and among various gene-editing technologies, CRISPR/Cas9 system is shown to be more precise and reliable. The aim of this study was to assess the possibility of knocking out exon 48 by using a pair of sgRNAs.
Methods: A pair of guide RNAs (gRNAs) was designed to cleave DMD gene and induce deletion of exon 48. gRNAs were transfected to the HEK-293 cell line and then the deletion in genomic DNA was analyzed by PCR and subsequent Sanger sequencing.
Results: Exon 48 was successfully deleted and therefore exon 47 was joined to exon 49.
Conclusion: This result indicated that CRISPR/Cas9 system could be used to edit DMD gene precisely.
Introduction :
Duchenne Muscular Dystrophy (DMD) is an X-linked recessive inherited neuromuscular disease which is caused by out of frame mutations in dystrophin gene (DMD) 1,2. DMD expresses a rod-shaped cytoplasmic protein, which is a member of dystrophin glycoprotein complex and has an important role in sarcolemma stability 3. In DMD patients, loss of dystrophin expression results in progressive muscle degeneration, loss of mobility, wasting of cardiac and respiratory muscle and premature death 2. DMD is the largest gene in human genome by 2.2 mega base pairs which is composed of 79 exons 4. A variety of mutations have been reported in DMD patients, but approximately 60-70% of them are deletions with most prevalent ones in hotspot region of exons 45 to 55 4. At present, there is no effective treatment for DMD patients and typical treatments such as corticosteroids and physical therapies only alleviate secondary symptoms of DMD 5. Knocking out mutated exons by CRISPR/Cas9 system induces production of a truncated but functional dystrophin protein 6. This approach would result in milder form of DMD that is Becker Muscular Dystrophy (BMD) 7. In contrast to DMD, in BMD, in frame mutations lead to DMD gene expression that synthesizes partially functional dystrophin proteins 7. The aim of this study was to assess the possibility of knocking out exon 48 by using a pair of sgRNAs.
Materials and Methods :
gRNAs design and cloning: According to DMD gene sequence (NG_012232. 1 RefSeqGene, NCBI), two gRNAs were designed (g47 and g48) and validated using CRISPR-MIT design tool (https://zlab.bio›guide-design-resources) to target exon 48 (Table 1). gRNAs were cloned into the backbone of the pSpCas9 (BB)-2A-GFP (PX458) using BbsI enzyme (Thermo Scientific). gRNA sequences are shown in table 1. The plasmid constructs were cloned into the Escherichia coli (E. coli) DH5a and were extracted using Miniprep kit (Qiagen).
Validation of gRNA insertion to the px458 vector: Single primer set was designed to detect correct insertion of gRNAs to the px458 vector (F: TTCTTGGGTAGTTTGCAGTTTTAA, R: CACGCGCTAAA AACGGACTA). PCR condition was set as initial denaturation at 95°C for 2 min, followed by 30 cycles of incubation at 95°C for 30 s, 54.6°C for 30 s, and 72°C for 30 s and last extension at 72°C for 5 min. PCR product was analyzed by Sanger sequencing (Figures 1, 2A and 2B).
Cell culture: Human embryonic kidney 293 cells (HEK 293 cells) (ATCC: CRL-1573) were cultured in high glucose DMEM (Dulbecco's Modified Eagle's Medium) that was supplemented with 10% FBS (Fetal Bovine Serum, Gibco) and 1% Penicillin-Streptomycin (Thermo Fisher Scientific).
Cell transfection: After 2 subsequent subcultures, cells were transferred to a 6 well cell culture plate and were seeded with Opti-MEM® I Reduced-Serum Medium (Thermo Fisher Scientific). After 24 hr, the HEK 293 cells at approximately 60% confluence were transfected by G47 and G48 using transfection agent TRANSFECTimin (Dara Zist-Fan Eram, Iran) according to the manufacturer’s instruction. After 8 hr of transfection, Opti-MEM medium was discarded, then full and fresh DMEM was added to the cells. After subsequent 48-72 hr, cells were monitored by fluorescence microscopy. Transfection efficiency was measured by eGFP expression in px458 constructs.
Fluorescence-activated cell sorting (FACS): Two days after transfection, GFP positive HEK 293 cells were trypsinized and collected for FACS, using FACSAria™ III (BD Bioscience). GFP positive cells were collected and expanded for molecular analysis.
Genomic extraction and PCR-based assays: Detection was carried out by designing two primers which flanked exon 47 and were called "DMD Forward" (TTCTTGGGTAGTTTGCAGTTTTAA) and "DMD Reverse" (CAGTAACAAATATGATGCAGG C) primers. Forward primer was located in intron 47 and reverse primer in intron 48. Genomic DNA was extracted (QIAGEN DNA extraction kit) and PCR condition was set as initial denaturation at 95°C for 2 min, followed by 30 cycles of incubation at 95°C for 30 s, 58°C for 30 s, and 72°C for 40 s, and last extension at 72°C for 5 min.
Results :
In this study, exon 48 was selected which is located in hotspot mutation region of DMD gene. Based on our study design, two target sites were chosen in introns 47 and 48. It was hypothesized that deletion of exon 48 would result in joining of exon 47 to exon 49 which would result in occurrence of an in frame deletion in this region. Eighteen hours after cloning of G47 and G48 guide RNAs in to the E. coli, DH5α white colonies appeared in LB agar plates that were supplemented with ampicillin.
Engineered colonies carrying gRNAs were detected by colony PCR and observation of the 241 bp PCR products on gel electrophoresis (Figure 2C). Sanger sequencing validated correct insertion of G47 and G48 gRNAs to the PX458 vector individually. After 72 hr of the cotransfection of the two gRNAs to the HEK-293 cell line, positive transfected cells expressed eGFP-Tag, and then GFP positive cells were sorted and collected (Figures 2D and 2E). Total genomic DNA was extracted from the cells to analyze the deletion of exon 48 in the edited cells. A pair of primers was designed outside the gRNAs target site and PCR reaction was conducted. In negative control cells, which were not transfected by any gRNAs, a 1400 bp product was amplified and detected on agarose gel electrophoresis. In addition to the 1400 bp band, a 700 bp product was amplified from total DNA extracted from positive transfected cells (Figure 2F). Sanger sequencing confirmed deletion of exon 48 and the joining of exon 47 to exon 49.
Discussion :
DMD has been recognized as a genetic disorder for over two decades and it affects 1:3500 male births. Common therapies only reduce the signs of the disease while there is not any curative treatment for DMD 2,8. Large size of DMD gene (2.2 M bp) is an obstacle for any gene editing technique; nonetheless, gene therapy has recently been considered a potential therapeutic option for DMD treatment 7.
Out of frame mutations in DMD gene are due to deletions, which usually occur in hotspot mutation region in exons 45 to 55 4. Prevalence of deletion mutations is different among various populations in the world. In Mexican-Mestizo patients, mutations in exon 51 are more prevalent 9; however, 78% of deletion mutations in Indonesian patients occur in exons 43 to 52 10. Exons 51 and 45 are reported as hotspots mutation exons in Spain 11. Among Iranian patients, DMD gene mutations diversify. Several studies used CRISPR/Cas9 for editing DMD gene. Min et al deleted exon 44 in cardiomyocytes differentiated from hiPSCs by CRISPR/Cas9 and restored dystrophin expression in the edited cells 12. In another study, Tabebordbar et al deleted exon 23 in mouse muscle and stem cells of mdx mice by CRISPR/Cas9 and they used AAV9 as vector for transfer of CRISPR system to the target cells 13. Their results indicated restoration of dystrophin in the edited cells in north west of Iran and 83% of deletions occur in exons 44 to 52 14. However, in south west of Iran, mutations are more frequent in exons 44 to 55 15. Considering the deletion prevalence pattern, particularly in Iranian DMD patients, exon 48 was selected in this study which is located in distal hotspot mutation region in DMD gene. Exon 48 was successfully deleted which consequently led to joining of exon 47 to exon 49. The result indicated that CRISPR/Cas9 system can edit DMD gene precisely. Although the use of CRISPR/ Cas9 as a therapeutic approach for DMD requires more vitro and in vivo studies, our efforts could be a small step toward achieving this goal.
Conclusion :
Our results indicate that successful deletion of exon 48 results in joining of exon 47 to exon 49, which could possibly lead to synthesis of partially functional dystrophin protein. Taken together, the results demonstrate that CRISPR/Cas9 is a powerful gene-editing tool, which could precisely edit dystrophin gene and could be regarded as a promising approach for DMD treatment in the future.
Acknowledgement :
The authors would like to thank the RCC center from Shiraz University of Medical Sciences for editing this manuscript.
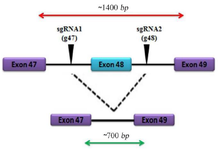
Figure 1. Schematic map of exon deletion in DMD gene by CRISPR/ Cas9 system.
|

Figure 2. Guide RNAs cloning to the PX458 vector using Sanger sequencing, A) Electropherogram of inserted gRNA G47 in PX458 vector, B) Electropherogram of inserted gRNA G48 in PX458 vector, C) Guide RNAs cloning to the PX458 vector using PCR and correct insertion of gRNAs to the px458 vector demonstrated a 241 bp band (M: DNA Marker 100 bp, S1-S5: Positive results, C-: Negative control), D) Light microscopy of the HEK-293 cell line, E) Florescent microscopy of GFP positive cells after transfection, F) Knocking out of exon 48 by PCR (M: DNA Marker 100 bp, C-: Negative control showing a 1400 bp band, S: 1400 and 700 bp bands in edited cells).
|

Table 1. gRNA sequences, based on DMD gene sequence, two gRNAs were designed (G47 and G48) to target exon 48
|
|