Retraction: The Frequency and Importance of Common α-globin Gene Deletions Among β-Thalassemia Carriers in an Iranian Population
-
 Moosavi, Azam
Department of Biochemistry, Faculty of Medicine, Alborz University of Medical Sciences, Alborz, Iran, Tel: +98 26 34336007, Fax: +98 26 34302090, E-mail: Azammoosavi1@gmail.com
Moosavi, Azam
Department of Biochemistry, Faculty of Medicine, Alborz University of Medical Sciences, Alborz, Iran, Tel: +98 26 34336007, Fax: +98 26 34302090, E-mail: Azammoosavi1@gmail.com
-
Department of Biochemistry, Faculty of Medicine, Alborz University of Medical Sciences, Alborz, Iran
-
Department of Molecular Medicine, Biotechnology Research Center, Pasteur Institute of Iran, Tehran, Iran
-
M. Ardekani, Ali
-
Department of Medical Biotechnology, National Institute of Genetic Engineering and Biotechnology, Tehran, Iran
Abstract: Background: β-thalassemia is the most common monogenic disorder in Iran, and one of the challenges in the screening of the carriers is the coinheritance of α-thalassemia mutations. In the view of high prevalence of α-thalassemia mutations in many parts of the country, the aim of this study was to determine the carrier frequency of common alpha deletions, as a secondary modifier in clinical manifestations of beta thalassemia, in known beta-thalassemia carriers and some hematology parameter changes.
Methods: The study included families referred from different primary health care centers with microcytic hypochromic anemia [MCV<80fl; MCH<27 pg] and A2>3.4%]. Genomic DNA was extracted from peripheral blood leukocytes by salting out method. For common β-globin gene mutation analysis, amplification refractory mutation system- polymerase chain reaction (ARMS-PCR) and for rare β-thal alleles, DNA sequencing were used. Also, for investigation of common α-globin gene cluster deletions (-α3.7, -α4.2, --MED and -α20.5), multiplex Gap-PCR was performed.
Results: Among 227 β-thalassemia minor individuals studied, α-globin gene deletions were found in 43 cases: 37 heterozygote -α3.7 (16.3%), 5 homo -α3.7 (2.2%) and 1 --MED (0.44%). Also, the co-inheritance of α-globin gene deletion and triplication was not found in the studied individuals.
Conclusion: Although it is highly recommended that physicians and genetic counselors involved in the screening program of beta-thal major in the country consider this phenomenon because of high prevalence of this coinheritance, hematologic indices changes are very slight.
Introduction :
The severity of β-thalassemia (β-thal) symptoms is associated with alpha and non α-globin chain ratio imbalance 1. It is expected that α-globin gene triplication/ quadruplication in a locus can aggravate the clinical phenotype of a defective β-globin gene, although that may be variable in different individuals in severity. On the other hand, deletion of α-globin genes in combination with beta-thal may ameliorate the clinical condition 2-5.
The national thalassemia screening program for prevention of β-thal major in Iran was initiated based on pre-marital screening since 1997 because of the high prevalence of β-thalassemia. Fetuses affected with thalassemia major can be aborted before 18 weeks of gestational age according to 1998 law permission 6.
The presence of different hemoglobin abnormalities in multiethnic community in Iran makes challenges in the screening program and genetic counseling 7-10. In this regard, the combination of α-globin numerical variants may change the blood parameters and clinical severity of β-thalassemia 11-13.
A single α-gene deletion co-inheritance effects on β0-thal are very slight, while a milder disease have been seen in β+ thal individuals with two α-gene deletions 2,14. Also, moderately severe anemia features are seen in Hb H disease (with only one functional α-gene) and homozygous β-thal patients 3,5. Although abnormal hematological parameters are not associated with α-globin gene triplication, there are some reports that indicate co-inheritance of α-globin gene triplication and heterozygous β-thal alleles might be responsible for β-thal intermedia phenotypes ranging from mild to blood transfusion dependent 2,15-17.
β-thal alleles average frequency is about 4-5% in Iran which could increase to 10% in northern and southern regions of the country while α-thal carriers frequency in some southern regions is about 30% 18,19. In addition, the carrier frequency of the αααanti3.7 triplication or quadruplication has been reported to be 2.14% in individuals with normal hematological indices and 1.7% in individuals with mild microcytosis 14.
In this study, the co-inheritance of β-thal alleles with common α-globin gene deletions was evaluated in asymptomatic carriers of β-thal in an Iranian population. Then, α-globin gene deletion cases were examined for α-globin triplication. Finally, the hematological indices and genetic consequences of this combination were considered.
Materials and Methods :
In this study, 227 unrelated β-thal carriers were referred from Primary Health Care (PHC) centers (from 2010-2012) to genetic prenatal diagnosis laboratories. All cases had low hematological indices (MCV <80 fL, MCH <27 pg and Hb A2 >3.4%).
Written informed consent was obtained from all participants in the study. Then, 5 ml peripheral blood was collected in tubes containing EDTA. DNA extracts were obtained from peripheral blood leukocytes by salting out method 20. For common β-globin gene mutation analysis, amplification refractory mutation system, polymerase chain reaction (ARMS-PCR) and for rare β-thal alleles, DNA sequencing was used. Also, for common α-globin gene cluster deletions (-α3.7, -α4.2, --MED and -α20.5), multiplex Gap-PCR based on Liu et al.’s study was performed 21. Also, to investigate alpha triplication (αααanti3.7) in cases with α-globin gene deletion, multiplex PCR method was set up and used 14.
Results :
In this study, the combination of α-globin gene deletion was investigated in an Iranian population of β-thal carriers referred from Primary Health Care centers. Out of the 227 unrelated asymptomatic β –thal carriers with defined mutation in HBB gene included in the study, 43 individuals (about 19 percent) had one or two α-globin gene deletions including 37 cases heterozygote for -α3.7 with αα/-α3.7 genotype, 5 cases homozygote for -α3.7 deletion with -α3.7/-α3.7 genotype, and one -MED deletion with αα/--MED genotype. The co-inheritance of α-globin gene deletion and triplication was not found in the studied individuals. In figure 1, a representative run of samples tested for α-globin gene deletions is shown. Also in table 1, important hematological indices were analyzed in beta thal screening in 2 groups.
Discussion :
Mediterranean, African and South Asian areas are more affected by beta-thalassemia. Approximately, Eastern Mediterranean, Europe, Western Pacific, Sub-Saharan Africa, Southeast Asia and America’s people are affected by a gene mutation at 2-18, 0-19, 0-13, 0-12, 0-11 and 0-3%, respectively 22,23. Rabbani in his review has pointed that, in beta globin gene there are more than 800 described variants worldwide which can result in beta thalassemia 24,25. In Sardinia (11-34%), Sicily (10%), Greece (5-15%), and Iran (4-10%), HBB mutations are seen frequently 26-30. In Iran, most common beta-thal mutations reported are IVS II-1 and IVS I-5 31.
Alpha thalassemia is a common single-gene disorder throughout parts of the world where malaria is endemic. Multi-ple studies have suggested that the presence of both single and double alpha-globin gene deletions has a protective ef-fect against malaria 32,33. The allele frequencies of alpha thalassemia in the Mediterranean basin, portions of West Afri-ca and in parts of Saudi Arabia, India, Thailand, Papua New Guinea, and Melanesia are 5-10, 20-30, and 60-80%, respectively. The frequency of heterozygote carrier status among the Chinese population has been reported to range from 5 to 15%. The frequency of alpha thalassemia is lower than 0.01% in Great Britain, Iceland, and Japan 34-36.
Although β-thalassemia results from reduced β hemoglobin synthesis, the amounts of free α-globin chains can be an important genetic modifier contributing to variation in hematological and clinical phenotypes in patients. The variabil-ity in clinical severity of β-thalassemia has been attributed to environmental and genetic factors, including α-triplication, α-deletion and Hereditary Persistence of Fetal Hemoglobin (HPFH), HFE variants and some other un-known mutations 1.
The alpha2 gene transcription amounts are two to three times more than alpha1 gene transcription in alpha gene cluster. So this different expression could result in hemoglobin variant in deletional and nondeletional forms of alpha-thalassemia 37. Alpha-thalassemia based on nondeletion defects happens in low frequencies. Point mutation or oligo-nucleotide deletions/insertions for alpha globin gene expression could be critical. The most common nondeletional are the T C initiation codon mutation, the -5nt alpha and -IVS1 deletion in Mediterranean and also polyadenylation site mutations in Mediterranean and Middle East populations, Hb Constant Spring and other resemblance elongated vari-ants in Mediterranean, middle East Asia, and Southeast Asia 38. MRNA product of these changes, because of disruption of the untranslated region seems as a reason of product reduction of Hb 39. Also, some mutations of alpha genes could produce hyper unstable globin variants that are unable to form stable tetramers. Harteveld et al have recently reported a complete nondeletional alpha-thalassemia mutation list 40.
In our study, the common α-globin gene cluster deletions (-α3.7, -α4.2, --MED and -α20.5) were investigated and -α20.5 deletion was not seen in our cases, while -α20.5 deletion was estimated to be about 13.6% in Qazvin province, 1.9 % in Gilan province and 1.5% in Mazandaran province 41-43. Also, in Khuzestan province -α20.5 deletion has not been detected 44. These results show that samples had Hb A1> 90%. Therefore, because of alpha mutation low occur-rences and our limitation for more investigation, the main focus was on alpha common deletions.
As shown in table 1, comparing means of individuals with alpha deletion in beta-thal carriers, alpha deletion can modify mean of indices in beta thalassemia carriers slightly. Means of Hb and MCV alteration were significant and in none of our samples, MCV was more than 80 fl. In MCH category, while our sample size was two times bigger but MCH mean increase in alpha-beta group was not significant. Our sample size for HbA2 and RBC indices was statically small but as seen in table 1, our results had very small differences.
It seems that our investigation results are different from other studies in the same field. It should be noted that in some of the reported studies, comparing is performed between alpha-beta thalassemia with normal cases or in more than two groups but our emphasis was comparison between beta thalassemia carriers with alpha- beta thalassemia carriers in hematology indices so increasing amounts occur in very small scales 8,45.
On the other hand, the frequency of α-triplication/quadruplication in some populations has been reported. In most cases, α-triplication occurs at no higher than about 1% of the world population though it has been observed in just over 3% of the Sri Lankan population. Both triplicated and quadruplicated α-globin-gene arrangements seem to have very little effect in otherwise normal individuals 1. It has been reported that a single additional α-gene had a very limited ef-fect on the beta-thal minor phenotypes or may produce the mild thalassemia intermedia phenotype with no history of blood transfusion 46. Based on these facts, all of β-thalassemia carriers that had α-globin genes deletion for alpha globin gene triplication/quadruplication were investigated to ensure that phenotypes of these cases are just related to β-thalassemia and α-globin genes deletion as genetic factors. The co-inheritance of α-globin gene deletion and triplication was not found in alpha-beta thalassemia studied individuals. Also, none of the affected persons with β-thalassemia and α-globin genes deletions showed higher hematological indices than fixed indices in β-thalassemia screening pro-gram in Iran.
Conclusion :
It seems that in such situations, the nature of HBB mutation, the number of α-globin genes and other environmental and genetic modifiers should be considered for genotype-phenotype correlation analysis. In our study, our findings indicate that although the β-thalassemia screening program in Iran is not special but it is sensitive enough in isolating people with β-thalassemia as a microcytic hypochromic anemia, even those cases with co-inheritance of common α-globin genes deletion.
At the present time, still many unclear points exist for prediction of the phenotype and it is confusing for genetic counselors to know exactly what is happening or what should be done to prevent the birth of children with thalassemia (legal and ethical aspects). Further studies are needed to clarify these questions especially in a multiethnic country like Iran.
Acknowledgement :
We thank all of our colleagues in the field of Molecular medicine at the Pasteur Institute of Iran who provided insight and expertise that greatly assisted the research. Also we appreciate the guidances provided by Dr. Masoud M.A. Boojar from Kharazmi University at the beginning of the study.
Conflict of Interest :
The authors declare that they have no conflict of interest.
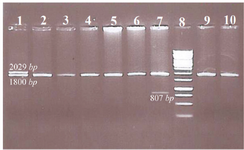
Figure 1. Representative samples tested for common α-globin gene deletions.
Well 1. Heterozygote for -α3.7 with αα/-α3.7 genotype
Well 2, 3, 4, 5, 6, 9 and 10. No of α-globin gene deletion, there are guest normal alpha alleles
Well 7. One -MED deletion with αα/-MED genotype
Well 8. Ladder
|
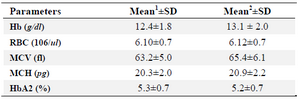
Table 1. The mean of hematological indices among beta-thalassemia and alpha-beta-thalassemia carriers
- The mean values with standard deviations for 100 beta-thal carriers without any common alpha deletion are shown in this column for specific parameters
- The mean values with standard deviations for 43 individuals with coinheritance of common alpha deletion and beta-thal mutation are shown in this column for specific parameters
|
|