Computational Detection of piRNA in Human Using Support Vector Machine
-
Seyeddokht, Atefeh
-
Department of Animal Science, Faculty of Agriculture, Ferdowsi University of Mashhad, Mashhad, Iran
-
 Aslaminejad, Ali Asghar
Department of Animal Science, Faculty of Agriculture, Ferdowsi University of Mashhad, Mashhad, Iran, Tel: +98 9155161513, E-mail: agr764@yahoo.co.uk
Aslaminejad, Ali Asghar
Department of Animal Science, Faculty of Agriculture, Ferdowsi University of Mashhad, Mashhad, Iran, Tel: +98 9155161513, E-mail: agr764@yahoo.co.uk
-
Department of Animal Science, Faculty of Agriculture, Ferdowsi University of Mashhad, Mashhad, Iran
-
Masoudi-Nejad, Ali
-
Laboratory of System Biology and Bioinformatics (LBB), Institute of Biochemistry and Biophysics, University of Tehran, Tehran, Iran
-
Nassiri, Mohammadreza
-
Department of Animal Science, Research Institute of Biotechnology, Ferdowsi University of Mashhad, Mashhad, Iran
-
Zahiri, Javad
-
Bioinformatics and Computational Omics. LAB (BioCOOL), Faculty of Biological Sciences, Tarbiat Modares University (TMU), Tehran, Iran
-
Sadeghi, Balal
-
Department of Food Hygiene and Public Health, Faculty of Veterinary Medicine, Shahid Bahonar University of Kerman, Kerman, Iran
Abstract: Background: Piwi-interacting RNAs (piRNAs) are small non-coding RNAs (ncRNAs), with a length of about 24-32 nucleotides, which have been discovered recently. These ncRNAs play an important role in germline development, transposon silencing, epigenetic regulation, protecting the genome from invasive transposable elements, and the pathophysiology of diseases such as cancer. piRNA identification is challenging due to the lack of conserved piRNA sequences and structural elements.
Methods: To detect piRNAs, an appropriate feature set, including 8 diverse feature groups to encode each RNA was applied. In addition, a Support Vector Machine (SVM) classifier was used with optimized parameters for RNA classification. According to the obtained results, the classification performance using the optimized feature subsets was much higher than the one in previously published studies.
Results: Our results revealed 98% accuracy, Mathew’ correlation coefficient of 98% and 99% specificity in discriminating piRNAs from the other RNAs. Also, the obtained results show that the proposed method outperforms its competitors.
Conclusion: In this paper, a prediction method was proposed to identify piRNA in human. Also, 48 heterogeneous features (sequence and structural features) were used to encode RNAs. To assess the performance of the method, a benchmark dataset containing 515 piRNAs and 1206 types of other RNAs was constructed. Our method reached the accuracy of 99% on the benchmark dataset. Also, our analysis revealed that the structural features are the most contributing features in piRNA prediction.
Introduction :
In recent years, numerous confirmations from high-throughput genomic programs show that albeit less than 2% of the mammalian genome translates proteins, a major segment can be transcribed into diverse mixed members of non-coding RNAs (ncRNAs) 1-3. The Encyclopedia of DNA Elements (ENCODE) and associated projects indicated that the majority of eukaryotic transcripts are ncRNAs 4. There are more than twenty thousand protein-coding genes in the human genome, which correspond to roughly two percent of human genome 5. The remaining regions in the human genome are non-coding RNAs, which were previously named "dark substance" or "junk DNA" 6. Recently, ncRNAs have attracted significant attentions with respect to their various biological roles, highlighting the biological importance of previously "overlooked" RNA reservoir 7. ncRNAs are complicated elements with different significant biological functions in the cell, including the control of chromosome dy-namics, RNA splicing, RNA excision, translational inhibition and mRNA demolition 8. ncRNAs can be coarsely categorized into small ncRNAs (such as small nucleolar RNAs (snoRNAs), short-interfering RNAs (siRNAs), piwi-interacting RNAs (piRNAs), microRNAs (miRNAs), and short hairpin RNAs (shRNAs)) or long ncRNAs (lncRNAs), based on the transcript size 9-12. The range of ncRNAs is growing rapidly as new ncRNAs remain to be discovered by high-throughput sequencing methods. Nevertheless, a large number of ncRNAs presumably cannot be recognized 8,13. Therefore, the identification and explanation of ncRNAs is a considerable step for the explanation of different regulatory mechanisms in the cell. piRNAs are about 19 to 33 nucleotides long and most of their sequences fall in the range of 25-33 nucleotides. These ncRNAs are similar to siRNA and miRNA which also have a strong preference for the 5’uridine. Besides, piRNA molecules are located in clusters of length 20-100 kb. The density of piRNA clusters ranges from 40 to 4000 14-18. piRNAs are the most diverse and the most expressed small ncRNAs in animals 19,20. They are involved in epigenetic and post-transcriptional regulation of retrotransposons 21. Lately, numerous researches have started to discover the hitherto unknown pathways of piRNA synthesis 22. Until now, a large number of piRNA sequences have been identified in human 23, mouse 16, rat 17, zebra fish 24, and fruit fly 25. Computational identification approaches can supply experimental approaches to identify ncRNAs quickly in novel genomes, specifically the ncRNAs that are transcribed under particular conditions in specific cell types. Many computational methods have been suggested for ncRNAs prediction, consisting of comparative 26-29 and non-comparative methods 30-37.
In recent years, some studies were devoted to analyze computational identification of piRNAs 38-41. Brayet et al 38 integrated machine learning method based on multiple kernels and a Support Vector Machine (SVM) classifier to identify the human and Drosophila piRNAs. Their method combined previously identified features and a new telomere/centromere neighborhood feature. The results from their SPG-GMKL method were better than the ones reported by Zhang et al 41 (>0.8 in almost all measurements for both Human and Drosophila). Wang et al 39 performed transposon interaction and a SVM for piRNAs prediction. They used SVM to predict human, mouse and rat piRNAs, and they achieved 90.6% accuracy. They developed Piano program to predict piRNAs for the rice stem borer, Chilo suppressalis. They achieved an accuracy and sensitivity of 95%, and 96%, respectively. Betel et al 40 trained a SVM classifier to distinguish between 5'-RNA and all other uridin positions for mouse piRNA sequences. In this way, they could identify mouse piRNAs with a precision of 61-72 percent. But their method could not effectively predict those piRNA derived from the 3'-UTR of mRNA which are produced by Ping Pong model. Also, Zhang et al 41 used Fisher separator algorithm by setting different cutoffs for piRNA identification in five model species including mice, humans, rats, fruit fly, and nematode. Their approach reached a precision of over 90% and a sensitivity of over 60%. But these studies are computationally intensive or they did not show a satisfactory prediction performance. In this study, to find a set of effective descriptors for discriminating piRNAs from other ncRNAs, heterogeneous features of ncRNAs were extracted 42,43. Then, the SVM classifier with different parameters was performed to detect an optimized feature subset. The results show that different feature types have different discriminative power in piRNA prediction. Also, according to the results (our method achieved accuracy of 98% and specificity of 99%), the proposed method can be used effectively for piRNA detection.
Materials and Methods :
Dataset: In order to acquire the appropriate piRNA dataset, 515 piRNAs from piRNABank 44 (http://pirnabank. ibab.ac.in/) as positive instances were extracted.
To construct negative dataset, three groups of RNAs were used. The first one was composed of precursor miRNA sequences (http://www.mirbase.org/, version 21), the second group consisted of non-piRNA sequences and they were derived from different databases including 1200 sequences of various types: sequences of lncRNA, extracted from NCBI (http:// www.ncbi.nlm.nih.gov). precursor miRNAs, extracted from miRBase (http:// www.mirbase.org/, version 21); and human mRNA sequences ,downloaded from the NCBI database (http://www.ncbi.nlm.nih.gov). And the third group was composed of:
sequences of snoRNA, collected from snoRNA-LBME-db database (https://www-snorna.biotoul.fr);
precursor miRNA sequences, extracted from miRBase (http://www.mirbase.org/, version 21); and sequences of tRNA, extracted from genomic tRNA database (http://gtrnadb.ucsc.edu/). As a result, 1206 sequences of the third group were selected as negative instances.
Extracted features
Sequence-based features: Sequence-based features have showed a discriminatory power to predict biological functions of macromolecules 45,46. With respect to the sequence features, the frequency of two neighboring bases (e.g., %AA), 15 sequence motifs 43 and the content of G and C (%G+C) formed the sequence-based feature sets.
Structural features: Generally, structural attributes are significant for the identification of human piRNAs. Thus, sequence and structural features were incorporated to recognize human piRNAs. RNAfold program 47 was used with the default parameters to calculate structural features based on the RNA secondary structures. Since the Minimum Free Energy (MFE) is an index that assesses the stability of the secondary structure of non-coding RNAs, several structural features, including MFE, as the structural feature sets were selected. Table 1 shows the total 48 features.
Support vector machine (SVM): SVM is an efficient machine learning technique, responding to classification problems in bioinformatics and computational biology 48. The SVM is able to convert low-dimensional non-linear matters into high-dimensional linear problems, resolving non-linear classification issues by reducing the linear classification problems 49. In this study, two models were trained and tested separately using one- and two-class SVM for human dataset. In addition, for optimizing the one-class SVM model, the radial basis function (RBF) kernel parameter nu (γ) were adjusted by the grid search strategy in MATLAB. Figure 1 illustrates the pipeline for piRNA identification.
Performance evaluation: A 10-fold cross validation procedure was used to assess the performance of the proposed model using four measures: sensitivity (SE), specificity (SPC), accuracy (ACC), and Matthews’s correlation coefficient (MCC). The formulas of these four measures are as follows:
SE=TP/(TP+FN)
SPC=TN/(TN+FP)
ACC=(TP+TN)/(TN+FP+TP+FN)
| MCC= |
(TP×TN-FP×FN) |
|
| √ (TP+FP)(TN+FN)(TP+FN)(TN+FP)
|
TP and TN are the number of piRNAs and non-piRNAs, respectively, that predicted correctly. Also, FN and FP are the number of piRNAs and non-piRNAs that predicted wrongly 38.
Results :
Kernel function selection: One of the most important parameters in SVM classifier is the kernel function 50. In this study, four different kernel functions were used to obtain the best SVM model for piRNA prediction: Linear, Polynomial, Radial Basis Function (RBF) and Sigmoid. As figure 2 shows, the SVM with RBF kernel performed better than the others.
Optimizing the nu (γ) parameter: Selecting the appropriate value for the nu (γ) parameter is a critical step in SVM model with RBF kernel function51. To optimize this parameter, different values of γ were tried (Figure 3). According to the obtained results, the best values for γ were 0.06 and 0.08 in the training dataset.
Assessing the prediction performance: The 48 features were divided into eight overlapping groups and in each group the optimized SVM was run. These eight groups were 15 motif features (Features 1-15), 10 features corresponding to the sequence (Features 16-25), 17 features corresponding to the sequence (Features 16-32), 9 structural features (Features 33-41), 7 features corresponding to the base pair (Features 42-48), 22 features corresponding to the motif and base pair (Features 1-15 and 42-48), 16 features corresponding to structure and base pair (Features 34-48) and finally, 31 features corresponding to structure, base pair, and motif (Features 1-15, and 33-48). Table 2 shows the performance of the SVM using different subset of features. As table 2 shows, the best performances were achieved for the fourth and fifth groups. Figure 4 provides a better illustration of the performance when different subsets of features were used.
Thus, such structural and base pair features (Table 2) that were previously used to classify real and pseudo miRNAs 42 propose the similarity of these structure-base pair features for all types of small ncRNAs 39.
Discussion :
In this paper, SVM was exploited to predict piRNA in human. After examining various kernel functions, RBF kernel function was used for the SVM. Choosing appropriate descriptors are of considerable importance to encode RNAs for building an accurate model. In this study, 48 various descriptors were used to build feature vectors. These features can roughly be categorized into eight overlapping groups (Table 2).
To assess the contribution of different feature types in piRNA prediction, performance of the SVM model was computed using different feature subsets. Our results showed that structural features (Features 33-41) and 7 features corresponding to the base pair (Features 42-48) are the most contributing features. These features had near perfect performance (accuracy of 98% and sensitivity of 99%).
Also, an attempt was made to compare the proposed method with the three recently published methods of Zhang et al 41, Lakshmi et al 43 and Brayet et al 38. Zhang et al 41 used k-mer schema to identify piRNA sequence in five model species. Liu et al 43 developed a method for piRNA identification based on motif discovery using SVM classifier, named Pibomd. Brayet et al 38 proposed an algorithm, named piRPred, to identify piRNAs. They used a multiple kernel fusion and an SVM-based approach which allow using heterogeneous features. To have a fair comparison, these methods were run on our dataset. As table 3 shows, our method outperformed the other methods in three different performance measures.
The biological significance and the functions of piRNA molecules are currently the subject of intensive study and, numerous researches have started to uncover the hitherto unknown biological mechanisms of piRNA 22,52,53. Therefore, computational identification of piRNA has been at the forefront of research for understanding the mechanisms that maintain germline integrity. According to the results, it can be stated that feature subsets 4 and 5 are the most contributing features in piRNAs prediction. So, secondary structure features and base pairing information, which can be computed by appropriate tools, can effectively help the biologists to design and discriminate piRNAs from other RNAs 47,54.
Conclusion :
In this paper, a prediction method was proposed to identify piRNA in human. Also, 48 heterogeneous features (sequence and structural features) were used to encode RNAs. To assess the performance of the method, a benchmark dataset containing 515 piRNAs and 1206 types of other RNAs was constructed. Our method reached the accuracy of 99% on the benchmark dataset. Also, our analysis revealed that the structural features are the most contributing features in piRNA prediction. Finally, three recently published computational studies were used in piRNA detection. The obtained results show that the proposed method outperforms its competitors.
Acknowledgement :
We are very grateful to Dr. Ahmadi and Dr. Azimzadeh, for their helpful suggestions.
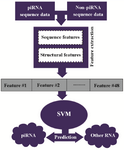
Figure 1. Flowchart describing the pipeline for piRNA identification.
|
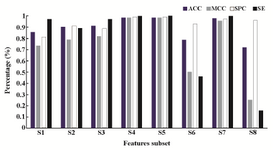
Figure 2. Different performance measures when different subsets of features were used.
|
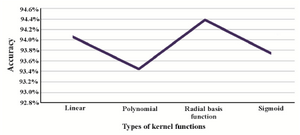
Figure 3. Accuracy of the SVM model with different kernel functions.
|
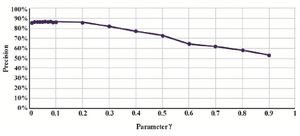
Figure 4. Different values of the parameter γ in SVM model.
|

Table 1. The final 48 features used for building our model
|
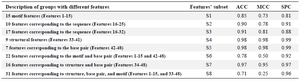
Table 2. Performance of the SVM using different subset of features
|
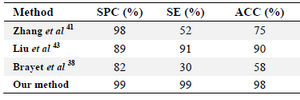
Table 3. Comparison with other methods
|
|