TP53 Binding to BRCA1 and RAD51 in MCF7 and MDA-MB-468 Breast Cancer Cell Lines In vivo and In vitro
-
 Rasti, Mozhgan
Recombinant Lab, Department of Biochemistry, Faculty of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran, Tel: +98 711 32303029; E-mail: rasti31@yahoo.com
Rasti, Mozhgan
Recombinant Lab, Department of Biochemistry, Faculty of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran, Tel: +98 711 32303029; E-mail: rasti31@yahoo.com
-
Recombinant Lab, Department of Biochemistry, Faculty of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
-
Azimi, Tayebeh
-
Recombinant Lab, Department of Biochemistry, Faculty of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
Abstract: Background: Tumour suppressor genes such as TP53, BRCA1 and RAD51 are involved in DNA repair and their malfunctions result in genomic instability and cancer. Wild type (WT) TP53 binds to BRCA1and RAD51 in vivo and in vitro. However, mutated TP53 in tumours can interfere with WT TP53 function. We studied how mutation of TP53 in MDA-MB-468 cell line could affect its binding capacity and interfere with WT TP53 interaction with these DNA repair proteins.
Methods: Binding capacity of mutated TP53 in MDA-MB-468 breast cancer cell line to BRCA1 and RAD51 proteins in comparison to WT TP53 in MCF7 cell line was studied by Immunoprecipitation. In vitro studies were performed by GST-WT p53 pull-down assays in these cell lines to assess the interaction of GST-WT p53 with BRCA1 and RAD51 proteins.
Results: The results showed that mutated TP53 in MDA-MB-468 cells interacted with BRCA1 protein in vivo and did not effect WT TP53 binding to this protein in vitro. The Immunoprecipitation assays revealed that the mutated TP53 did not bind to RAD51 in comparison to WT TP53. However, this mutated protein could not interfere with binding of RAD51 to GST-WT p53 in MDA-MB-468 cell line by in vitro experiment.
Conclusion: It was found that WT TP53 interactions with BRCA1 and RAD51 did not interfere with mutated TP53 in MDA-MB-468 cell line. In addition, RAD51 did not bind to TP53 with R273C mutation in vivo.
Introduction :
Breast cancer is a world-wide cancer in women. In Iranian women especially the age of its diagnosis has decreased recently 1. Breast cancer genome analyses have shown that there are a few tumour suppressor genes that are frequently mutated such as breast cancer 1, early onset (BRCA1) and tumour protein p53 (TP53) genes 2. TP53 is mutated in the majority of sporadic breast cancers. Most of these cancer related mutations in TP53 are in their DNA binding domain that reveal a gain-of-function for TP53 3. In such cases TP53 mutated levels are increased in breast tumours and can interfere with repair of DNA double strand breaks 4,5. In normal tissue only a small amount of TP53 is present that binds to DNA to induce cell cycle check point activation, cellular apoptosis, autophagy and senescence 6. BRCA1 mutation is associated with hereditary breast and ovarian cancers 2. It binds to Mre11-Rad50-NBS1 (MRN) complex which is a DNA repair complex for DNA double stranded break and ZBRK1, a zinc finger transcriptional repressor 7. In addition, it regulates cell cycle check points 8. On the other hand, BRCA1 interacts with RAD51 recombinase (RAD51) during homologous recombination mediated DNA double-strand break repair forming a nucleoprotein filament on DNA and its mutation can increase the risk of breast cancer 9-11.
It has been shown that Wild Type (WT) TP53 is able to bind both to BRCA1 and RAD51 to conduct genomic stability 12,13. However, mutated TP53 in tumours not only interferes with WT TP53 function but also impair chemotherapy responses 14. In the present study, our aims are to study how a mutation of TP53 protein in its DNA binding domain can affect its binding capacity and interfere with WT TP53 binding to these DNA repair proteins. Therefore, we evaluated the binding capacity of WT TP53 to these two proteins in MCF7 and MDA-MB-468 cell lines in comparison to mutated TP53 in MDA-MB-468 breast cancer cell line in vivo and in vitro. Such studies can elucidate the molecular mechanism of mutated TP53 in comparison to wild type one in order to provide novel diagnostic and therapeutic insights.
Materials and Methods :
Cell culture: Both MDA-MB-468 cells which possess a mutation in codon 273 of the TP53 gene and MCF7 cells with WT TP53 gene were used in this study (Pasture Institute, Iran). These cancer cell lines were grown in RPMI 1640 (Biosera, UK) supplemented with L-glutamine to 2 mM, and 10% fetal calf serum (Cinagen, Iran) and sub cultured 2-3 times in a week.
In vitro experiment by GST pull-down assay: The p53-Glutathione S-transferase (GST) fusion protein was expressed and purified as described previously 15. The MCF7 and MDA-MB-468 cell lysates were solubilized in a lysis buffer containing 10 mM Tris-HCl pH=7.4, 0.825 M NaCl and 1% NP-40 and then sonicated and cleared by centrifugation. All protein concentrations were measured by Bradford protein assay and concentrations of samples were calculated based on standard curve 16. For GST pull-down assay 17, 50 µg of GST- WT p53 or GST proteins was mixed with 5 mg of either the MCF7 or MDA-MB-468 cell lysates for 2 hr at 4oC. The pull- downed complexes with GST and GST-WT p53 proteins were eluted from the glutathione-agarose by 25 mM reduced glutathione in 50 mM Tris-HCl solution pH=8.0. The eluted samples were resolved by Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then subjected to Western blot analysis with appropriate antibody.
In vivo experiment by immunoprecipitation of proteins: The studied cancer cells were lysed in buffer and protein concentrations were measured as described above. Immunocomplexes were precipitated from 5 mg of protein lysate 17, using 5 µg of primary antibodies of anti-BRCA1 (SC-641; Santa Cruz, USA) or anti-TP53 serum 15. IgG Ab was used as negative control. After 2 hr mixing at 4oC on a rotator, 30 µl of packed protein G agarose beads (Sigma, UK) was added into the protein-antibody complex and mixed for a further 1 hr. Immunocomplexes bound to the beads were then spun and washed, prior to re-suspending in the SDS sample buffer for SDS-PAGE analysis.
Western blot analyses: GST pull-down or immunoprecipitated samples with 50 µg proteins of appropriate lysates were electrophoresed on 8 and 12% SDS-PAGE gels for detection of BRCA1 and RAD51, TP53 proteins, respectively. GST pull-down complexes were immunebloted with 1:100 dilution of anti-BRCA1 (SC-641; Santa Cruz, USA) and 1:1000 dilution of anti-RAD51 (ab63801; Abcam, USA) antibodies. In addition, anti-TP53 serum 15 and anti-RAD51 (ab63801; Abcam, USA) primary antibodies were diluted 1:1000 for analyzing of immunoprecipitated proteins in blots. A secondary antibody of horseradish peroxidase-conjugated anti-rabbit (Sigma, USA) with dilution of 1:1000 and chemiluminescence substrates (ECL; Amersham Bioscience AB) were used to detect the immuno-labeled bands.
Results :
TP53 mutation in MDA-MB-468 cell line did not affect its binding capacity to BRCA1: In order to understand the binding capacity of mutated TP53 in MDA-MB-468 cell line in comparison to wild type one in MCF7 cells, cell lysates of these cells were immunoprecipitated with antibody against BRCA1 and blotted for TP53. The results (Figure 1A) showed that both mutated and WT p53 proteins were able to bind with this repair protein. GST pull-down assays were performed with GST-WT p53 fusion protein synthesized in Escherichia coli (E. coli) and these lysates to analyze by BRCA1 immunoblotting. In vitro results (Figure 1B) revealed that GST-WT p53 bound to BRCA1 and mutated TP53 in MDA-MB-468 cell line did not interfere with binding of the wild type fusion protein to BRCA1. BRCA1 did not bind to GST alone showing there was no binding affinity between GST alone and BRCA1. Figure 1B shows that the expression of BRCA1 in MDA-MB-468 cell line was more than MCF7 cell line that provided more proteins for binding to GST-WT p53 in MDA-MB-468 cell line rather than MCF7 cells. Therefore, co-precipitation of GST-WT p53 with BRCA1 in MCF7 cell line was underrepresented as judged from autoradiograph.
Mutated TP53 in MDA-MB-468 cell line was not able to bind RAD51: Immunoprecipitation of TP53 in MDA-MB-468 and MCF7 cell lysates and blotting for RAD51 revealed that mutated TP53 in MDA-MB-468 cell line was not able to bind RAD51. However, this in vivo experiment for MCF7 cell line showed that the precipitated immunocomplex included WT TP53 and RAD51 (Figure 2A). Interaction of WT p53 to RAD51 in MCF7 cell lysate was confirmed by pulling down of GST- WT p53 fusion protein with RAD51and blotting for RAD51 in two studied cell lines. Figure 2B shows that GST-WT p53 pulled down the RAD51 proteins in these two cell lines. However, mutated TP53 was not able to bind the RAD51 in MDA-MB-468 cells, the mutated TP53 did not interfere with binding of GST- WT P53 to RAD51 in this cell line. RAD51 did not bind to GST alone that showed there was not binding affinity between GST alone and RAD51.
Discussion :
WT TP53 is stabilized and accumulated in normal cells due to the cellular stress and DNA damage. However, mutant alleles of TP53 are highly expressed in one third of breast tumors 14. In addition, it has been shown that mutant p53 proteins are able to inhibit WT TP53 through dominant negative gain of function. Different mutated p53 proteins may inhibit DNA damage response, cell cycle control and apoptosis process induced by the WT one. Therefore, mutated forms can drive tumor formation, invasion and metastasis that impair chemotherapy responses which are based on the WT TP53 function 14. MDA-MB-468 cell line is hemizygous for a mutated p53 gene which makes a R273C mutant and ectopic expression of WT TP53 in these cells cannot overcome the gain of function of mutant form 18. This mutant TP53 protein is reported to cooperate with oncogenes for transforming cells and inhibits p73 protein 14.
Genetic instability is another result of TP53 mutation that is because of inhibition of DNA repair pathway. Some of TP53 mutants can bind to MNR complex and impair recruiting taxia-telangiectasia-mutated (ATM) kinase to double strand break 19. WT TP53 binds to BRCA1 and RAD51 to induce repair pathways in the case of any DNA damage 12,13. Many studies showed that how BRCA1 is involved in DNA repair and cell cycle check points to maintain genome stability 2,7,8,20. BRCA1 binds to RAD51 9 which possesses a main role in Homologous Recombination (HR) by repairing endogenous and exogenous DNA double-strand breaks 11. It happens during DNA replication and uses sister chromatid as a template to repair the DNA double-strand break 21.
Our in vitro experiments using GST- WT p53 protein in studied cell lines confirmed previous studies 13 that WT TP53 binds to RAD51 directly. We found that this interaction was not interfered by the mutated TP53 in MDA-MB-468 cell line. These data shows that RAD51 interacted with bacterial extracted GST- p53 protein and did not need post transcriptional modification of p53 for its binding. It has been reported that RAD51physicaly interacts to GST-WT p53 and to a lesser extent a point mutant GST-273H-p53 protein. Mapping studies revealed that RAD51 binds two regions of amino acids 94-164 and 264-315 of TP53 13. In addition, our in vivo result for RAD51 in MCF7 cell line is consistent with previous data in other cell lines 22 and confirmed that it was able to bind WT TP53.
In the present study, for the first time it was shown that RAD51 protein failed to interact with mutated TP53 in MDA-MB-468 cell line in vivo. It reveals that despite the previous in vitro results for 273H-p53 point mutant with reduced amount of binding to RAD51 13, R273C mutant diminished interaction activity completely in vivo. It implies a main role for this point mutant in TP53 for binding to RAD51. This finding can explain the genomic instability in MDA-MB-468 cell line that may result in the harsh metastasis property of these cells. It has been reported that BRCA1 binds to C-terminal domain of TP53 between amino acid residues 300-393 12.
Our in vivo experiments showed that BRCA1 was over expressed and precipitated with mutated TP53 in MDA-MB-468 cell line which indicated its point mutation in 273 codon did not effect its binding affinity to BRCA1. However, because low levels of BRCA1 were present in MCF7 cell line we could not show this interaction with WT TP53. This interaction has been revealed in other cell line previously 12. The in vitro results with GST-WT p53 confirmed previous finding for direct interaction of WT-p53 with BRCA1 12 and showed that mutated TP53 in MDA-MB-468 cell line could not interfere with its interaction to BRCA1. It has been shown that BRCA1 is a potent co-activator of TP53 in p21 and bax genes transcription 12. In MCF7 cells, TP53 is maintained at very low level by proteasome degradation when there is not any cellular stress. In MDA-MB-468 cell line, there is genomic instability that activates mainly ATM and related proteins 23. While ATM activation stabilizes TP53 and BRCA1 by phosphorylation 23, it may result in the increased expression levels of the mutated TP53 and BRCA1 that we observed in MDA-MB-468 cell line.
Conclusion :
We found that BRCA1 protein but not RAD51 interacted to TP53 with R273C mutant in vivo and this point mutant in TP53 did not interfere with the binding capacity of WT TP53 to BRCA1 and RAD51.
Acknowledgement :
This work was supported by grant No: 88-01-01-1395 from Shiraz University of Medical Sciences, Iran for Ms. Tayebe azimi’s thesis. The authors would like to thank the School of Advanced Medical Sciences and Technologies for their support in performing this project.
Conflict of Interest :
None of authors has any conflict of interest.
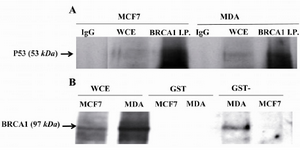
Figure 1. TP53 bound to BRCA1 in vivo and in vitro. A) In vivo binding of wild type (WT) TP53 in MCF7 cell line and mutated TP53 in MDA-MB-468 (MDA) to BRCA1. Cell lysates were immuno-precipitated with Ab against BRCA1 and IgG and then the immuno-complexes were Western blotted for anti-p53. B) Mutated TP53 did not interfere with in vitro binding of WT TP53 to BRCA1. BRCA1 was pulled down in MDA-MB-468 (MDA) and MCF7 cell lysates by GST-WT p53 that was a fusion protein synthesized in prokaryotes cells and analyzed by Western blotting with anti-BRCA1. WCE whole-cell extract, I.P. Immunoprecipitate.
|
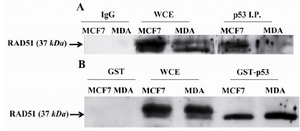
Figure 2. RAD51 only bound to wild type (WT) TP53 in vivo and in vitro. A) RAD51 bound to WT TP53 in MCF7 cell line but not to the mutated one in MDA-MB-468 (MDA) cells. Cell lysates were Immunoprecipitated with Ab against TP53 and IgG then analyzed by Western blotting with anti-RAD51. B) Mutated TP53 did not interfere with in vitro binding of WT TP53 to RAD51. RAD51 was pulled down in MDA-MB-468 (MDA) and MCF7 cell lysates by GST-WT p53 that was a fusion protein synthesized in prokaryotes cells to assay the binding capacity of RAD51 to synthetized TP53. Western blot analysis was performed with anti-RAD51. WCE whole-cell extract, I.P. Immunoprecipitate.
|
|