Comparative Analysis of Prostate Cancer Gene Regulatory Networks via Hub Type Variation
-
Khosravi, Pegah
-
Department of Bioinformatics, Institute of Biochemistry and Biophysics (IBB), University of Tehran, Tehran, Iran
-
School of Biological Sciences, Institute for Research in Fundamental Sciences (IPM), Tehran, Iran
-
Akbarzadeh, Mohammad
-
Department of Bioinformatics, Institute of Biochemistry and Biophysics (IBB), University of Tehran, Tehran, Iran
-
Mirkhalaf , Samira
-
Department of Bioinformatics, Institute of Biochemistry and Biophysics (IBB), University of Tehran, Tehran, Iran
-
Sadeghi, Mehdi
-
School of Biological Sciences, Institute for Research in Fundamental Sciences (IPM), Tehran, Iran
-
National Institute of Genetic Engineering and Biotechnology (NIGEB), Tehran, Iran
-
 Goliaei, Bahram
Department of Bioinformatics, Institute of Biochemistry and Biophysics (IBB), University of Tehran, Tehran, Iran, Tel: +98 21 66498672, Email: bahram.goliaei@gmail.com
Goliaei, Bahram
Department of Bioinformatics, Institute of Biochemistry and Biophysics (IBB), University of Tehran, Tehran, Iran, Tel: +98 21 66498672, Email: bahram.goliaei@gmail.com
-
Department of Bioinformatics, Institute of Biochemistry and Biophysics (IBB), University of Tehran, Tehran, Iran
Abstract: Background: Prostate cancer is one of the most widespread cancers in men and is fundamentally a genetic disease. Identifying regulators in cancer using novel systems biology approaches will potentially lead to new insight into this disease. It was sought to address this by inferring gene regulatory networks (GRNs). Moreover, dynamical analysis of GRNs can explain how regulators change among different conditions, such as cancer subtypes.
Methods: In our approach, independent gene regulatory networks from each prostate state were reconstructed using one of the current state-of-art reverse engineering approaches. Next, crucial genes involved in this cancer were highlighted by analyzing each network individually and also in comparison with each other.
Results: In this paper, a novel network-based approach was introduced to find critical transcription factors involved in prostate cancer. The results led to detection of 38 essential transcription factors based on hub type variation. Additionally, experimental evidence was found for 29 of them as well as 9 new transcription factors.
Conclusion: The results showed that dynamical analysis of biological networks may provide useful information to gain better understanding of the cell.
Introduction :
The complexity and multigenic nature of cancer has necessitated various genome-wide studies to achieve a systems-level understanding of the key genetic mediators involved in prostate cancer 1. Most diseases are due to the collapse of cellular processes together with interaction networks 2. Therefore, exploring the biological network for complex diseases provides an understanding of the functional alterations in chronic diseases 3.
Network-based approaches contain many clinical applications to explore human diseases systematically. A better understanding of the effects of cellular networks on disease progression may lead to the identification of disease genes which, in turn, may offer better targets for drug development 4. One focal point in cancer analysis is the reconstruction of Gene Regulatory Networks (GRN) 5. However, cancer progression is a dynamic process with multiple stages; so, reconstruction of one static GRN may not be informative enough for the inference. Instead, reconstructing stage-specific GRNs during cancer progression and then comparing these GRNs would be beneficial to characterize the main genes and interactions involved in cancer progression.
The availability of genome-wide gene expression data has helped develop various state-of-art GRN reconstruction methods 5-7. These methods seek to identify putative gene regulatory interactions by assuming that alterations in the expression level of a regulator (such as a transcription factor) have a direct effect on the cognate regulated genes.
Empirical evidence of extensive GRN rewiring during cancer progression, along with the availability of GRN reverse engineering approaches, have inspired us to conduct a systematic investigation to characterize the topological changes that occur in a prostate cell’s GRN during cancer progression. Therefore, in this study, an attempt was made to reveal candidate disease-associated genes and biomarkers for prostate cancer progression by integrative gene expression profiling and network analysis at a systematic level. In this way, four stage-specific GRNs were reconstructed based on a comprehensive prostate cancer gene expression dataset containing 171 different samples monitoring gene expression at different disease phases. Topological comparison of these four GRNs based on hub type variations recapitulates the previous findings about extensive GRN rewiring 8. Through sub-network analysis, it is possible to identify significant genes which were supposed to change their hub type with highly relevant to specific phases of prostate cancer.
Enormous efforts have been made to identify biomarkers for various cancers by the analysis of different transcriptome data 9-11. Moreover, there were similar studies for analysis of sub-networks or hub genes which had been helpful for the understanding of the metastasis of cancer at the molecular level 12. Nonetheless, there are still few studies on identification of prostate cancer biomarkers for disease progression 13. Therefore, in this study, a new integrative network-based approach was developed to detect party hubs and date hubs based on Degree and BN algorithms during cancer progression.
Our analysis led to identification of 38 important genes putatively involved in prostate cancer. Through extensive literature search, experimental evidences revealed the role of 76.3% of candidate genes in prostate cancer (Table 1). This level of experimental confirmation reflects the high accuracy of the proposed approach.
Our study hereby demonstrates a useful approach for analysis of prostate cancer at the systematic level. For the genome-wide investigations, this will be a fundamental attempt for future development of the translational medical informatics, which lead to better patient diagnostics with high-throughput data through systems biology 14.
Materials and Methods :
Network reverse engineering approaches: Reverse engineering of GRNs from whole genome data entails deciphering the underlying gene regulatory circuits by observing changes in gene expression profiles 5. With advances in high-throughput technologies, several computational reverse engineering approaches using different statistical measures have been developed 15-18, including information-theoretic network inference methods, which identify connections between genes by approximating the quantity of information common to any pair of genes. In the Dialogue on Reverse Engineering Assessment and Methods 5 (DREAM5) challenge, the context likelihood relevance (CLR) algorithm by Faith et al 6 had the best performance among information theory based approaches 19.
Briefly, CLR determines an interaction between two genes to be significant by estimating the significance of their Mutual Information (MI) value against a background distribution of the MI values of every other pair involving one of the two genes of interest. In this way, the significance level is dynamically determined for each interacting pair according to their expression profiles. Given a gene expression dataset and the significance scores calculated by CLR algorithm, the corresponding empirical False Discovery Rate (FDR) can be estimated by running the algorithm on randomly shuffled datasets. In this study, all GRNs were reconstructed using CLR with an FDR threshold of 0.05. It is important to note that CLR relies solely on the dependency between expression profiles to detect interactions. Consequently, the resulting network is a co-expression network; so, the GRN is extracted from this network by considering only interactions where at least one transcription factor is involved.
Prostate cancer microarray data: Prostate cancer microarray data were downloaded from the Gene Expression Omnibus (GEO) database, accession number GSE6919 20. This dataset contains 171 samples, including samples from normal prostate tissue free of any pathology (Normal with 18 samples), normal prostate tissue adjacent to tumors (Adjacent with 63 samples), primary prostate tumor tissue (Tumor with 65 samples), and metastatic prostate cancer (Metastasis with 25 samples). Microarray data were preprocessed and analyzed using the LIMMA package in R 21 which was originally developed for differential expression analysis of microarray data. Quantile normalization and a moderated t-statistic were used to find differentially expressed genes. More detailed descriptions of the methods can be found in the original publications.
Network topological analysis: To predict the key regulators in the prostate cancer based on hub type variations, the stage-specific GRNs of prostate cancer were searched for transcription factors which either had a high number of connections or were bottleneck 22,23. The bottleneck genes are important because if they are removed from a network, the network will be disrupted, as they are major intersections between clusters in the network 24. To find such genes, all constructed GRNs were topologically analyzed following the same rules using cyto-Hubba package 25 which is a plugin for one of the most useful structural analysis software, Cytoscape 26. Cyto-Hubba is used to detect the critical nodes of biological networks with many topological algorithms such as Degree, Bottleneck (BN), Maximum Neighborhood Component (MNC), Density of Maximum Neighborhood Component (DMNC), and a Double Screening Scheme (DSS) 25-27. As mentioned before, Degree and BN were used to find the top ranked genes in all GRNs based on hub type variation which was the ultimate goal in this research.
Results :
Stage specific network reconstruction of prostate cancer: Using the CLR algorithm, four independent networks related to the four different cell stages were reconstructed (Normal, Adjacent, Tumor, and Metastasis). The metastasis GRN had the lowest number of interactions with 2505 interactions while the other three GRNs had around 3000 interactions each. Additionally, topological analysis of the GRNs revealed that all four networks exhibited the small-word property 28 and scales-free architecture 29 which are the well-known characteristics of most biological networks (Figure 1). All four reconstructed GRNs were mainly composed of the same set of genes; however, the conserved interactions among these four networks were very low and the metastasis network had the most unique interactions (Figure 2).
Detection of essential transcription factors involved in the prostate cancer: Considering the importance of hub and bottleneck proteins in the structure of GRNs, the 50 highest-ranked genes were identified for each stage-specific GRN based on their degree and bottleneck scores, separately. Top 50 genes were selected based on previous studies that showed the highest percentage of critical proteins found in top 50 ones based on Degree and BN algorithm 25. Although there were four conditions that resulted in detection of 200 genes, 144 of 200 genes had overlap during various conditions. Consequently, 56 unique candidate genes were selected for further analysis.
In each GRN, these 56 genes were categorized based on their degree and bottleneck scores in four groups: 1) Hub-NonBottleneck: genes with high degrees and low bottleneck scores are putative party hubs 30; 2) Hub-Bottleneck: genes with high degrees and high bottleneck scores are putative date hubs 30; 3) NonHub-Bottleneck: genes with low degrees and high bottleneck scores; 4) NonHub-NonBottleneck: genes with low degrees and low bottleneck scores.
The results showed hub type variation for 38 genes across different stages, whereas 18 other genes were functionally conserved as date hubs under all conditions (Table 2).
Sub-networks consisting of the first neighborhoods of the 38 critical bottleneck transcription factors were extracted and compared, revealing changes in the number of interactions and gene targets between the stages. For some transcription factors such as STAT1, AR, HLF, ZEB1, TCF21, ISL1, KLF6, HOXB13, SIM2 and FOXA1, the number of interactions in the metastasis stage decreased dramatically (Table 3); the interaction numbers of other transcription factors, such as ZNF529, FOXC1, MNX1, and JUNB, increased considerably in the metastasis stage, indicating network rewiring (Table 3). These 14 transcription factors (Figure 2) showed dramatic changes in their number of interactions (fold change ≥2) during the cancer progression (Table 3).
Discussion :
To reconstruct cell stage specific GRNs, an attempt was made to focus on the available comprehensive transcriptome dataset, originally published 20. This dataset was generated by sampling from four different types of prostate tissues including normal cells (Normal), normal cells adjacent to cancer cells (Adjacent), primary tumor cells (Tumor), and metastatic cells (Metastasis). In our approach, genes were analyzed and prioritized based on the transcriptome data. Hence, it was possible to make reliable predictions only for genes with altered expression level across various conditions. To focus on these genes, only up/down-regulated genes were considered (fold change ≥2 and p<0.05) in at least one cell stage (978 genes). Also, enrichment of known cancer genes was checked among this set by using a previously curated list of 555 high confidence cancer genes, originally published 31. 100 genes were collected and identified as mediators in metastatic prostate cancer from 32, and 276 genes were added and annotated as either a cancer pathway or prostate cancer gene in the KEGG database. It was found that the cancer-related genes were about 1.55-fold (hypergeometric two-tailed test, p=2.52E-6) and the prostate cancer-related genes were about 2.22-fold enriched (hypergeometric two-tailed test, p=6.07E-6) in our selected gene set which were fluctuated during prostate cancer.
To identify master regulators and their associated circuits governing cell-specific behavior in each state, the GRNs of prostate cells were compared in different stages with each other. Because the CLR algorithm merely relies on the similarity of expression patterns to infer interactions, constructed networks in this step contain both regulatory interactions (interactions between regulated genes and their putative regulators) as well as interactions between co-regulated genes (non-regulatory interactions). Hereafter, these networks are called co-expression networks. To extract gene regulatory interactions from these networks, only interactions involved at least one human transcription factor were considered and a list of them were extracted 33,34. These networks are referred to as GRNs.
To predict the key genes in the prostate cancer, an attempt was made to find the stage-specific co-expression networks of prostate cancer for high connectivity (hub) or bottleneck genes 22,23. Hub and bottleneck properties are considered important centrality indices because they are major intersections between clusters in the network and if they are removed from a network, the network will be disrupted 24. Han et al suggested the existence of two types of protein hubs in the protein-protein interaction networks, namely party hubs and date hubs 30. Although both interact with many proteins, the difference is that party hubs are proteins that interact with many other proteins simultaneously, whereas date hubs interact with their partners asynchronously 30. By definition, the bottleneck proteins are responsible for the interconnection of clusters in the network, and thus bottlenecks with high degrees are most likely to be date hubs which contain groups of genes that assist in presenting common functions 24,35. The obtained results recapitulate previous findings in which some active sub-networks contained regulatory interactions were supplanted by new interactions which changed their degrees during different conditions 36.
The result also reflected the high level of rewiring of gene regulatory circuits during cancer progression, as suggested elsewhere 8. As shown in table 3, for example, more than 2-fold decrease in the number of interactions for KLF6 was observed which controlled cell cycle progression and apoptosis. Indeed, experimental data suggest that KLF6 is inactivated in many cancers such as prostate, ovary and colon 37,38. On the other hand, consistent with more than 2-fold increase in the number of interactions (from 14 in normal stage to 33 in the metastasis stage) for FOXC1 (Table 3), it was indicated that this gene is linked to androgen dependent growth of prostate cancer 39.
Our result led to identification of 38 transcription factors which were bottleneck and changed their interaction during cancer progression. Although the functional role of some famous transcription factors such as AR, SMAD3 and VDR are well known as genes linked to prostate cancer 40-42, the 9 transcription factors (CAMTA1, ISL1, MNX1, NHLH2, NKX2-2, STAT2, ZNF146, ZNF205, and ZNF529) are new candidates that may have critical roles in prostate cancer based on topological significance and regulatory changes during cancer progression. Among the remaining 9 transcription factors, 5 of them were associated with other cancer types. ZNF146, CAMTA1, NKX2-2, MNX1, and ISL1 are most prominent in colorectal cancer, neuroblastoma, Ewing's sarcoma, leukemia and breast cancer, and bladder cancers, respectively 43-48. No evidence could be found to show the relationship between the 4 remaining transcription factors and any type of cancer.
Conclusion :
In this paper, an accurate network-based framework for the analysis of transcriptome data was presented. The analysis of prostate state specific GRNs revealed 38 transcription factors which are critically important for prostate cancer progression. Also, 14 transcription factors were identified to be linked putatively to prostate cancer metastasis stage, so they would be used as key factors for future research in the field of cancer studies. Additionally, experimental evidences revealed the role of 29 of candidate transcription factors in prostate cancer.
The low number of predictions and high degree of overlap with previously known events in the prostate cancer demonstrate the high efficiency of our approach. In addition, the low number of predicted gene sets makes it easy to design follow up experiments to validate the results. In this study, it is believed that the results may provide critical information to gain better understanding of networks’ dynamics in the cell through complex diseases such as cancer.
Acknowledgement :
Pegah Khosravi has been supported by the School of Biological Sciences of Institute for Research in Fundamental Sciences (IPM). Vahid H. Gazestani has been supported by CIHR Systems Biology Fellowship. The authors would like to thank Dr. Gary Bader and Dr. Juri Reimand in the Bader’s lab for their invaluable insights.

Figure 1. The architecture of gene regulatory networks. All four networks (normal, adjacent, tumor and metastasis networks) follow the well-known characteristics of most biological networks; A) scale-free architecture defined as few highly connected genes (hubs) that link the other less connected genes to the network; B) small-word property which means any two genes in the network can be connected by relatively short paths through all interactions.
|
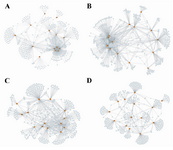
Figure 2. Number of interactions. This figure shows the GRNs for 14 TFs (orange nodes) that change their interaction numbers dramatically during cancer progression; A) Normal stage; B) Adjacent stage; C) Tumor stage; D) Metastasis stage which reflects the high level of rewiring of gene regulatory interactions.
|

Table 1. The function of 29 critical transcription factors putatively involved in prostate cancer
|
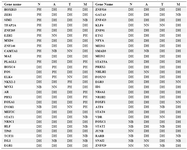
Table 2. 56 transcription factors showed different topological characteristics in different stages
N: Normal, A: Adjacent, T: Tumor, M: Metastasis, DH: Date Hub, PH: Party Hub, NB: Nonhub-Bottleneck, NN: Nonhub-Nonbottleneck
|

Table 3. The number of interactions for 14 out of 38 transcription
factors changed dramatically from normal to metastasis stage (fold change >2)
|
|