Computational Survey of FHIT, A Putative Human Tumor Suppressor, Truncates Structure
-
Ghahremani, Mohammad Hossein
-
Department of Pharmacology-Toxicology, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran
-
 Sardari, Soroush
Drug Design and Bioinformatics Unit, Medical Biotechnology Department, Biotechnology Research Center, Pasteur Institute of Iran, Tehran, Iran, Tel: +98 21 66405535; Email: ssardari@hotmail.com
Sardari, Soroush
Drug Design and Bioinformatics Unit, Medical Biotechnology Department, Biotechnology Research Center, Pasteur Institute of Iran, Tehran, Iran, Tel: +98 21 66405535; Email: ssardari@hotmail.com
-
Drug Design and Bioinformatics Unit, Medical Biotechnology Department, Biotechnology Research Center, Pasteur Institute of Iran, Tehran, Iran, Tehran, Iran
Abstract: Background: Fragile Histidine Triad protein (FHIT), as a known tumor suppressor protein, has been proposed to play crucial role in inhibiting p53 degradation by MDM2. Studies have confirmed FHIT interaction with p53 or MDM2, although functional interacting domains of FHIT with MDM2 and/or p53 are not completely defined. Thus, through determining the significant structural interacting domains of FHIT, information with regard to MDM2 and p53 would be provided. As there were no previous studies evaluating the interaction of optimized important parts of target molecules, docking study was employed.
Methods: Truncated structures of FHIT were screened to reveal critical sections engaging in FHIT interaction. HEX program was used in order to study the interaction of target structures.
Results: Given the total energy, FHIT structures (β5-7, α1) and (α1) of FHIT were showed to be better candidates in comparison with other structures in interaction with optimized MDM2 part. Furthermore, FHIT structures (β4-7, α1) and (β5-7, α1) were considered to be better than other structures in interaction with optimized p53 part. FHIT truncates which interact with MDM2 optimized part exhibited lower energy levels than FHIT truncates which interact with p53 optimized part.
Conclusion: Our results can be useful for designing new inhibitors of this protein complex interaction which would result in tumor repression.
Introduction :
FHIT belongs to Histidine Triad (HIT) nucleotide-binding protein superfamily and is considered a tumor suppressor 1. Genomic alterations and aberrant expression of FHIT have been correlated with many types of human cancers, including those of the lung 2-4, breast 5,6, cervix 7, colon 8, pancreas 9, prostate 10, stomach 11,12, head and neck 13.
Studies have shown the interaction of FHIT with MDM2 and the block of the interaction of MDM2 with p53, result in increased stability of p53 14. Structurally, FHIT forms a dimer in solution (PDB code: 1FIT) and general structure of its protomer can be described as a common α+β type 15. An ordinary hydrophobic core is formed within the background of the dimer 16.
As prior studies demonstrate, MDM2 protein interacts with p53 directly 17,18 and MDM2 can interact with FHIT (by immunoprecipitation) 14. Moreover, other studies confirm the interaction of p53 and FHIT 14,19. Thus, it is logical to consider that FHIT and p53 have binding sites on MDM2 and perhaps these proteins could influence each other in binding to MDM2.
As functional interacting domains of FHIT with MDM2 and/or p53 are not completely defined, therefore, by exploring the recognition site of interaction of FHIT-MDM2 with regard to p53 binding, one can evaluate the interaction and/or competition amid these proteins for therapeutical approaches. Furthermore, the research for functional domain of FHIT may reveal the protein domain responsible for tumor suppression. In addition, these studies will shed lights on the molecular mechanism of FHIT-MDM2-p53 complex.
In this study, we assessed FHIT constructs interaction with MDM2 and p53 optimized models in silico. Results can be useful for designing new inhibitors of this protein complex interaction which would result in tumor repression.
Materials and Methods :
Tertiary structure determination of FHIT truncates: PDB file of FHIT consisted of residues 2-106 and 127-147, was achieved from protein databank (PDB code: 1FIT). Based on previous studies, fourteen segmented structures were constructed by truncating pdb file using ViewerLite42 (2010).
PDB file of MDM2 and p53 optimized truncates, consisted of amino acids 23-119 of MDM2 and 18-26 of p53, were achieved from protein databank (PDB code: 1T4F) with using ViewrLite42.
FHIT, MDM2 and p53 modeling:The structure prediction process consisted of sequence alignment, model building, and structure refinement stages 20.
The tertiary structure of full FHIT was established by homology modeling using the pdb three dimensional structure of FHIT, available from the protein databank (1FIT) as template by Swiss homology modeling server, and Modeller 9v7 (2009) package (Table 1) 21.
Homology modeling was used to achieve the complete models of MDM2 and p53. Swiss homology modeling server and I-TASSER server were used for modeling the gaps of MDM2 and p53. Connecting MDM2 and p53 five segments was performed by means of Modeller 9v7 program. Ramachandran plot generated by spdb viewer was used to assess the best models of each molecule. Nominees were evaluated using PROCHECK. Energy minimization of the obtained structures was performed using Swiss-Pdb Viewer 4.01 (2010).
Computational interaction studies: Docked conformations and interaction energies were achieved using the protein-protein docking package HEX 5.1 (2008). During docking operation by HEX, the free energies were estimated based on shape complementarity only and shape/ electrostatics. The mean computational time used for a complex was about 30 min for HEX. HEX was executed on an IBM attuned computer running at 4 GB RAM and 2.5 GHz Dual Core™2 Intel® CPU.
Results :
Determination and generation of FHIT truncated structure:As mentioned before, we created 14 different truncated structures (Table 1), namely 2 to 12b and compared them with full length FHIT (Structure 1, Table 1) to recognize the most critical regions of FHIT involved in the interaction with MDM2 and p53 and also evaluate trimeric interaction complex. Tertiary structures of MDM2 and p53 optimized truncates, composed of amino acids 23-119 and 18-26, were obtained from protein databank (PDB code: 1T4F) (Figure 1).
Complete FHIT, MDM2 and p53 models:FHIT (Structure 1, Table 1), MDM2 and p53 structures were generated by homology modeling (Figure 2).
Interaction analysis:HEX results: The docking results performed by HEX for FHIT truncates with complete MDM2 and MDM2 optimized part, p53 optimized part are shown in tables 2-4.
In table 2, E-totals refer to the interaction of FHIT constructs with partial MDM2 of 1T4F (cut p53 optimized part) and p53 optimized part of 1T4F (deleted MDM2 part).
Table 5 indicates docking total energy of complete MDM2 and p53 as well as MDM2 optimized part and p53 optimized part.
Considering the shape and electrostatic energies, FHIT truncated forms 9 (β5-7, α1), 9b (β5-7, α1) and 11 (α1) interact with MDM2 optimized part at lower total energy and the interaction of FHIT with p53 optimized part is better for structures 6 (β4-7, α1), 9 (β5-7, α1) and 5 (β3-7, α1) based on total energy (Table 2).
Table 5 demonstrates total interaction energy of optimized part of MDM2 with optimized part of p53 obtained from PDB protein databank. Docking interaction energy of these two structures is -407.20 (kJ/mol). This table also shows total interaction energy of MDM2 complete model and p53 complete model
(-399.25 kJ/mol).
As table 3 shows, truncated structures 7 (β4-7, α1), 6 (β4-7, α1) and 4 (β1-5) interact with MDM2 complete model with lower E-shape (shape energy). Truncated structures 4 (β1-5), 5 (β3-7, α1) and 7 (β4-7, α1) dock with MDM2 complete model at lower total energy (shape and electrostatic energy) when compared to others. In general, structures in lower E-shape results have lower E-total (E-shape plus E-force) results.
Since there is a probability that FHIT and p53 might interact with MDM2 in a competitive way, we have also examined the interaction of triple protein complex FHIT, MDM2 optimized model and p53 optimized part in two stages. For this analysis, after interaction of FHIT constructs with optimized part of MDM2, these complexes (FHIT truncates with MDM2 optimized part) were tested with optimized part of p53. Similarly, following the interaction of FHIT truncates with optimized part of p53, the interaction of these complexes with optimized part of MDM2 was performed.
As table 4 illustrates, the complexes of truncates 2 (β1-2), 9 (β5-7, α1), 11 (α1) and MDM2 optimized part interact with p53 optimized part in higher total energy status. On the other hand, complexes of p53 optimized part with truncates 12 (β6-7), 11 (α1) and 12b (β6-7) interact with MDM2 optimized part in a higher total energy circumstance. Figure 3 represents three dimensional view of some FHIT, MDM2 optimized part, and p53 optimized part interaction view. Figure 4 represents two dimensional view of C9 (FHIT34-106), MDM2 optimized part, and p53 optimized part important interactions view.
Discussion :
Three dimensional structures of proteins are essential for computational interaction research. As only parts of FHIT, MDM2 and p53, had been determined as three dimensional structures in protein databank, we performed modeling for their structures.
Docking is considered as an in silico method for investigating the best interaction between two molecules which can be used in the rational drug design 22. HEX program actually accelerates the process compared with the classical FFT docking algorithms 23. Earlier studies have proved that HEX method has no limitation for protein size 24.
We tested docking of MDM2, p53 optimized parts as the receptor to compare the interaction tendencies of a special motif or a group of them within FHIT. The results of our previous docking study indicate that interaction of full FHIT with p53 (E-total: -568.66) and MDM2 (E-total: -459.53) is associated with lower total energy compared to the interaction of the complete MDM2 with p53 (E-total: -399.25). The abovementioned interaction occurred with higher total energy in comparison with the optimized p53 and MDM2 (E-total: -407.20). Moreover, subsequent to MDM2 and P53 optimization, it appeared that their relative tendency was augmented compared to their corresponding complete models.
Given the interaction of full FHIT with optimized models of p53 and MDM2, it is evident that FHIT truncates have higher affinity to interact with MDM2 optimized part than p53 optimized model. According to the interaction values, FHIT truncates interact with optimized MDM2 at lower E-total than optimized part of p53 and the total energies of docking interactions are directly related to shape energies of the mentioned interactions. Our results revealed that the tendency of β4-7, α1 segment of FHIT to p53 optimized model is more than other parts. Likewise, the β5-7, α1 structure of FHIT has more affinity to MDM2 optimized part than other forms. Thus, one can suggest the β5-7, α1 segment of FHIT as an interacting domain for both p53 and MDM2.
Having studied the above mentioned interactions, we found that FHIT remarkably has better affinity to bind MDM2 optimized part in the presence of p53 and MDM2 optimized models in most of the cases. Even though it can bind to p53 optimized model with low energy, when MDM2 optimized part is added to the model, the interaction with p53 optimized part is further attenuated (Table 4).
Interestingly, complex of FHIT truncates interacting with MDM2 optimized part at lower total energy usually interact with p53 optimized model at higher total energy. Thus, these findings indicate a sequence/ conformation specificity of FHIT truncates for interacting with MDM2 or p53.
E-totals of interaction between MDM2 optimized model (for interaction with p53) and FHIT truncates reveal that α1 is important in these interactions. E-totals of interaction between p53 optimized model (for interaction with MDM2) and FHIT truncates show that β5-7 and α1 are important parts in these interactions.
Experimental reports using yeast two-hybrid 25 and immunoprecipitation indicate that p53 at amino acids 1-41 25 or 1-52 26 interacts with MDM2. On MDM2, the interaction at amino acids 1-118 25 or 19-102 26 is the binding site to p53. Site-directed mutagenesis confirms the Leu14, Phe19, Leu22, and Trp23 of p53 are essential amino acids for interaction 27. The co-crystal structure of MDM2-p53 complex shows that Phe19, Trp23, and Leu26 are three main interacting residues in p53 28.
MDM2 directly binds to p53 18 and regulates p53 function and degradation 29-33. Moreover, a number of studies reported p53 and FHIT interaction 14,19 and their possible association 34. As shown in table 3, in p53-MDM2 interaction, the significant part of p53 is amino acids 18-26, and the important part of MDM2 is amino acids 23-119 35. Based on our results, the interaction sites of FHIT with MDM2 and p53 have overlapping parts. The best interaction site for MDM2-FHIT is residues 34-106 containing β5-7, α1. Conversely, for FHIT-p53 interaction, amino acids 21-104 containing β4-7, α1 are involved. However, residues 34-104 are involved in both interactions (Table 2). Interestingly, when the interaction of FHIT with MDM2 optimized part is challenged with p53 optimized part, the interaction site is different from interaction with MDM2 optimized part alone. Based upon these results, the interaction sites of FHIT with MDM2 and p53 are different with overlapping parts. FHIT binds to MDM2 with lower energy in the presence of p53 and the binding site shifts toward FHIT-MDM2 interaction. These data provide information involving competition of FHIT with p53 in binding to MDM2. Then, in the presence of FHIT, p53 is released from MDM2 and can increase apoptosis or cell cycle arrest. Figures 3 and 4 confirm the results indicated in the tables.
Conclusion :
In conclusion, our findings provide valuable information to understand the molecular mechanism of FHIT-MDM2-p53 complex formation and the design of inhibitory compounds. The truncated parts of FHIT with higher absolute E-total energy interacting with MDM2 optimized part [parts (β5-7, α1) and/or (α1)] can be effective in inhibiting degradation of p53 through altering MDM2 interaction with p53. Also, the truncated parts of FHIT with higher absolute E-total energy interacting with p53 optimized part [parts (β5-7, α1) and (β4-7, α1)] can be effective in inhibiting degradation of p53 through altering MDM2 interaction site with p53.
Acknowledgement :
This work was supported by Drug Design and Bioinformatics Unit, Pasteur Institute of Iran.

Figure 1. MDM2 optimized part (left), p53 optimized part (between) and 1T4F (PDB file) (right)
|
Figure 2. MDM2 complete model (left), p53 complete model (between) and FHIT complete model (right)
|
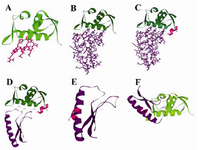
Figure 3. FHIT truncates, MDM2 optimized part, and p53 optimized part interactions three dimensional view. A) Three dimensional representation of 1T4F (PDB file); B) C9 FHIT truncate, MDM2 optimized part interaction view; C and D) C9 FHIT truncate and MDM2 optimized part interaction complex challenged with p53 optimized part; E) C9 FHIT truncate and p53 optimized part interaction view; F) C9 FHIT truncate and p53 optimized part interaction complex challenged with MDM2 optimized part
|
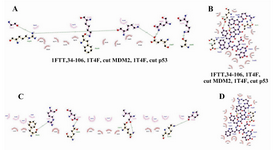
Figure 4. FHIT truncates, MDM2 optimized part, and p53 optimized part interactions, two dimensional view. A, B) C9 FHIT truncate and p53 optimized part interaction complex challenged with MDM2 optimized part; A) five residues of C9 involve in hydrogen bond in interaction with MDM2 optimized part, in this complex; B) p53 optimized part binding pocket. This binding pocket is composed of 17 residues in which three of them involve in hydrogen bond (Arg18, Asp21, and Trp23). C and D) C9 FHIT truncate and MDM2 optimized part interaction complex challenged with p53 optimized part; C) four residues of C9 involve in hydrogen bond in interaction with MDM2 optimized part, in this complex; D) p53 binding pocket. This binding pocket is composed of 14 residues in which one of them involve in hydrogen bond (Arg18)
|
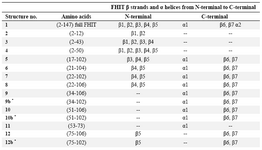
Table 1. FHIT truncated structures
* Note: C9b, C10b, C12b were created by truncating 4 amino acids from the end point of each constructs
|
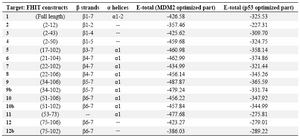
Table 2. Docking interaction energies (kJ/mol) of FHIT truncates with MDM2 and p53 optimized part
|
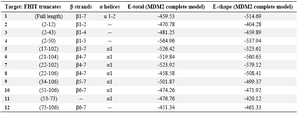
Table 3. Docking interaction energies (kJ/mol) of FHIT truncates with MDM2 complete model
|
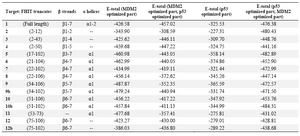
Table 4. Docking interaction energies (kJ/mol) of MDM2 optimized part with FHIT truncates, interaction of this complex with p53 optimized part, interaction of p53 optimized part with FHIT truncates, and interaction of this complex with MDM2 optimized part
|

Table 5. Docking interaction energies (kJ/mol) of MDM2 model with p53 model and optimized part of MDM2 with optimized part of p53
MDM2 complete model (1-484) and optimized part (Glu23-Val119); p53 complete model (1-392) and optimized part (Arg18-Leu26)
|
|