Retraction: Genotyping Analysis of Circulating Fetal Cells Reveals High Frequency of Vanishing Twin Following Transfer of Multiple Embryos
-
 Mouawia, Hussein
Department of Biology, Faculty of Science, Lebanese University, Beirut, Lebanon, +96 13786535; hussein_mouawia@yahoo.fr
Mouawia, Hussein
Department of Biology, Faculty of Science, Lebanese University, Beirut, Lebanon, +96 13786535; hussein_mouawia@yahoo.fr
-
Department of Biology, Faculty of Science, Lebanese University, Beirut, Lebanon
-
Paris Descartes University , Paris, France
Abstract: Background: Detection of Circulating Fetal Trophoblastic Cells (CFTC) by single cell genotyping not only allows to identify fetal cells from maternal blood, but also to characterize their bi-parental genome.
Methods: We have tested intact fetal trophoblastes recovered at 4th to 10th weeks of gestation (WG) from blood (10 ml per mother) of 13 women after In Vitro Fertilization (IVF) and transfer of one or several embryos. Large cells isolated from blood were individually microdissected and studied by genetic fingerprinting with a mean number of 3 Short Tandem Repeats (STR) markers, known to be informative by testing paternal and maternal blood DNA.
Results: CFTC were found in all mothers starting from the 5th WG. A mean number of 2.5 CFTC per ml of blood was found in all the analyzed samples collected at the different terms of pregnancy. All mothers who received the transfer of two or three embryos, including one who delivered twins and one with vanishing twin (identified by ultrasounds), were found to have CFTC with two or three different bi-parental genotypes, belonging to different embryos derived from the same parents.
Conclusion: CFTC circulation is detectable starting from the 5th WG. A "vanishing twin" phenomenon frequently develops after IVF and transfer of multiple embryos, being undetectable by ultrasounds and revealed by genetic CFTC fingerprinting.
Introduction :
Non-invasive Prenatal genetic Diagnosis (NI-PND) is a long-standing goal to avoid miscarriage related to invasive prenatal diagnostic procedures 1. Initial advances involved recovery of intact fetal cells, usually erythroblasts, from maternal blood 2-5. The Fluorescence Activated Cell Sorting (FACS) and Magnetic Activated Cell Sorting (MACS) approaches used to isolate circulating fetal erythroid cells resulted in 74% sensitivity in detecting trisomy 21 6. However, the approach only targeted pregnant women carrying a male fetus and consistently obtaining results has not been possible 6, thus precluding clinical introduction. Other approaches have since been pursued and current emphasis has shifted to cell-free fetal DNA 7. However, the amount of free fetal DNA in plasma is highly variable, usually a low proportion of maternal free DNA (3.4-6.2%) 8. Furthermore, a mixture of fetal DNA with large majority of maternal DNA makes detection of fetal abnormalities with NI-PND technically complex. Thus, recovery of intact fetal cells still retains unique potential to develop a reliable NI-PND method.
Circulating fetal cells include lymphoid, myeloid precursors, erythroid cells and epithelial (trophoblastic) cells. Among them, trophoblastic cells are suitable targets for non invasive prenatal diagnosis of the ongoing pregnancy while lymphoid and myeloid precursors are known to persist in the maternal blood for several decades after delivery 9. We observed that CFTC can be found in the blood of mothers at 10-11 weeks of gestation, making their use for NI-PND especially attractive 10-13.
The method we have developed allows identifying CFTC in blood through the detection of the inherited maternal and paternal allele in individually microdissected cells. Furthermore, it also allows characterizing the CFTC genome by DNA fingerprinting. In fact, when the genome of single microdissected cells is studied by STR genotyping with multiple informative markers, the two inherited paternal and maternal alleles are displayed and the fetal cell genome can be characterized for previous NI PND.
The present work has been planned in order to 1) determine the mean frequency of CFTC in maternal peripheral blood at different term of pregnancy during the first trimester, and 2) characterize the genome of CFTC found in mothers undergoing IVF after transfer of multiple embryos.
The goal was to assess the possibility of CFTC recovery providing a precise early non-invasive prenatal diagnostic test after the transfer of multiple embryos in mother and study the relevance of the existence of different CFTC in blood of the same mother who had a singleton pregnancy.
Materials and Methods :
Mothers: We studied 13 women who underwent successful IVF. Four women underwent the transfer of 3 embryos, 8 of 2 embryos and 1 of 1 embryo. All women delivered a singleton child except one who delivered twins (mother No. 2, Table 1). Mother No. 6 (Table 1) had a vanishing twin identified by ultrasounds at the 6th WG. Blood samples were collected from the 4th to the 12th WG. Among the 13 mothers, 6 underwent IVF for the first time and 2 had a previous IVF more than one year before. Among the remaining 5 mothers, No. 3 and 2 had previous IVF or Intra Uterine Insemination (IUI) 7 and 3 months before this study, respectively (Table 1). For the other 3 mothers, there are no data concerning their previous IVF.
Methods: A blood sample was obtained weekly from the 4th to the 12th WG from thirteen women (mean age: 38 years) who conceived by IVF, the goal being to determine the number of CFTC per ml, and per gestational age. The 13 women undergoing IVF were recruited in the Necker Hospital (Paris-France). Ten ml of maternal blood and 1 ml of paternal blood were collected in ethylenediaminetetraacetic acid (EDTA) buffer. Paternal and maternal DNA were extracted from 1 ml of blood and 1.5 ng was used for allelotyping with fluoresceinated primers specific for Short Tandem Repeat (STR) markers (D7S480, D7S486, D7S490 and D7S523, D16S539, D16S3018, D21S1435 and D21S1437).
The remaining 9 mL of maternal blood was treated by filtration on porous membrane up to 3 hr after collection, as previously described, filters were then stored at -20°C. After immunohistochemical analysis with KL1 antibody 13 to identify epithelial cells; cells were permeabilized with 0.2% Triton for 10 min before immunostaining. Primary antibodies were diluted 1:100 in 10% fetal calf serum and applied to the spot for 1 hr at room temperature. We used KL1 (Cytokeratin gp 56 kd; Immunotech S.A., Marseille, France), a cytokeratin broad-spectrum monoclonal antibody; anti-placental alkaline phosphatase (DAKO, Glostrup, Denmark), a monoclonal antibody for the evaluation of many different types of germ cells; and anti-leukocyte common antigen (DAKO), a monoclonal antibody recognizing a family of high-molecular mass glycoproteins expressed on the surface of the majority of human leukocytes. The following negative controls were performed: 1) the procedure was performed omitting the primary antibody; 2) the primary antibody was substituted by an irrelevant antibody (anti-HPV, B580; DAKO). As a positive control, we used fetal cells dissociated from human placenta, resuspended in the filtration buffer, and filtered.
Single cell laser microdissection was performed using laser-equipped microscope. Epithelial cells were microdissected using a Nikon microscope with MMI equipment and software (Zurich, Switzerland). The filter is then placed in the microscope with cells facing downward; the laser directly cuts the filter around the cell of interest to be microdissected. The back of the filter then adheres to the center of the underside of the lid of the Nikon tube, making it possible to expose the lysis buffer to the cell. To target epithelial cells for laser microdissection, we used assessment of cell size by MMI (Molecular Machines & Industries, Glattbrugg, Switzerland) CellCut software and filter calibrated pore size as a reference. A variable volume of blood was analyzed per WG (Table 2).
Each microdissected cell was lysed in 15 µL of lysis buffer (100 mmol/L Tris-HCl, pH=8; 400 µg/mL proteinase K) for 2 hr at 60°C, followed by proteinase K inactivation at 94°C for 15 min. For Primer Extension Preamplification (PEP) -14 to the lysed cell we added 5 µL of a 400 µM solution of random primers (Kit genPEPtm 75 OD, Genetix, Boston, USA), 6 µL of PCR buffer (25 mM MgCl2/gelatin (1 mg/mL), 100 mM tris-Hcl, ph8.3, 500 Mm KCL), 3 µL of a mixture of 4 dNTPs (each at 2 mM) and 1 µL (5 U) of Taq polymerase (Applied Biosystem, Foster City, CA, USA) in a final volume of 60 µL. STR amplifications were performed in 60 µL containing 6 µL of the PEP product, 10 mM Tris-HCl, 50 mM KCl, 2.5 mM MgCl2, 200 µM of each deoxynucleotide, 0.5 µM of each "outer" primer and 2 U of Taq Gold (Applied Biosystems, Foster City, CA, USA). Two µL of the 1:10 diluted PCR product were re-amplified in 20 µL final volume using "inner" fluoresceinated STR primers and the same PCR protocol. One µL of the 1:20 diluted inner PCR product was then mixed with 13.5 µL of deionized Hi-Di formamide and 0.5 µL of Genescan 400 HD (ROX) marker (Applied Biosystems) and loaded into an ABI Prism 3100 automated sequencer (Applied Biosystems). Profiles were analyzed using the Genescan and Genotyper software programs (Applied Biosystems).
Identification of CFTC was performed by amplifying, in parallel and using the same STR primers, 1.5 ng of PBL-derived paternal DNA and/or 1.5 ng of PBL-derived maternal DNA.
Controls of specificity: In addition, a negative control (buffer without sampling) was inserted for each sample at the lysis step and run to the end of the test. When performing laser microdissection, we always included at least one microdissection from a new filter (without cells) which was run in parallel with samples and controls.
Results :
We studied a total number of 106 filters and microdissected a mean number of 7 cells to identify one CFTC (total No. microdissected cells: 1946 cells). We performed 5232 single cell genotyping analyses allowing obtaining results with two or more informative STR markers per microdissected cell (CFTC DNA fingerprinting).
Determination of the earliest term of pregnancy at which CFTC are detectable: Among the 8 mothers tested at the 4th WG, only 4 of them were found to have detectable and very rare CFTCs (mean frequency: 0.2 CFTC/ml). CFTC were detectable at the 5th WG in all the 13th mothers tested (Table 2).
Determination of the mean frequency of CFTC in maternal peripheral blood at different term of pregnancy during the first trimester: Overall, 264 CFTC were identified in the 13 pregnant women by single cells microdissection and DNA fingerprinting, thus a mean number of 20 CFTC per mother. We analyzed a mean number of 8 blood samples per mother and found a mean number of 2.5 CFTC per sample. This result is reported in table 2, where pooled data from WG 6 to 8 and 9 to 12 are displaying the number of CFTC per mother and per sample.
Thus, the identification of CFTC by genetic analysis allowed establishing that 1) all the tested mothers had detectable CFTC starting from the 5th WG and 2) the frequency of CFTC (2.5 per ml) was not significantly different at the different terms of pregnancy.
Genetic characterization of the genome of CFTC found in mothers undergoing IVF after transfer of multiple embryos: Figure 1 shows an example of informative STR marker applied to the paternal and maternal blood DNA, to the DNA of the born baby and to large KL1 positive cells identified in the maternal blood. Paternal alleles are arbitrarily named a and b and maternal alleles are named c and d. In some case the maternal blood contains CFTCs displaying a bi-parental identical genome (a, c) as compared to that of the born baby (and thus belonging to the born baby) and CFTCs having a bi-parental DNA (b, c) different from that of the born baby.
All 8 mothers who received the transfer of two embryos (mothers No. 4, 5, 6, 7, 9, 10, 11, 12), including one who delivered twins (mother No. 2) and one with a vanishing twin (identified by ultrasound) (mother No. 6), were found to have CFTC with two different bi-parental genotypes.
Among the 4 mothers who received the transfer of 3 embryos (mothers No. 1, 2, 8, 13), one displayed CFTC with three different genotypes (mother No. 8), including the genotype of the born child, two displayed CFTC with two different genotypes (mothers No. 2 and 13), including one (mother No. 13) having CFTC with the genotype identified in the born child, and one (mother No. 1) displayed CFTC with only one genotype.
Finally, mother No. 3, who received the transfer of only one embryo displayed CFTC with only one genotype.
Among the 11 mothers having CFTC with different bi-parental genotypes, 4 were affected by primary sterility and underwent their first IVF (No.1, 3, 6, 7, 10, 11), two women had a previous IVF 8 years (No. 5) and 19 months (No. 8) before the current IVF. Among the remaining 5 mothers, three had previous IVF or ART 7 months before (No. 4, 12 and 13), and two (No. 2 and No. 9) had IVF or ART 3 months before the current IVF.
Mothers number 5, 6, 7, 8, 10 and 11 underwent IVF for the first time or had a previous IVF more than one year before, and received transfer of 2 (No. 5, 6, 7, 10 and 11) and 3 (No. 8) embryos, which indicates that the CFTCs having different bi-parental genotypes found in their blood belong to the transferred embryos.
We could obtain cells (mouth swab) from the born children of 8 mothers (No. 5, 7, 8, 9, 10, 11, 12, 13). The DNA extracted from these cells was genotyped with the same markers used for CFTC fingerprinting and allowed to demonstrate, in all the 8 cases, that one CFTC subtype (arbitrarily referred to as a/c in Table 2) belong to the born child. The CFTC with the different bi-parental genotype (referred to as b/c in Table 2) were thus predictably belonging to the other transferred embryos.
When we looked at the frequency of CFTC derived from the embryo who survived and gave a born child, we found that the frequency of this CFTC subtype was not substantially different at the different terms of pregnancy (1st trimester) (Table 2), confirming the finding that CFTCs start to circulate at the 5th WG and their frequency remains approximately unchanged from the 5th to the 12th WG.
Overall, by analyzing CFTC from the 5th to the 12th WG, we found CFTC with two different bi-parental genotypes in 10 of 12 mothers who received the transfer of two or three embryos. CFTC with 3 different bi-parental genotypes were found in one mother who received 3 embryos, and CFTC with 1 genotype were found in one mother who received only one embryo, 1 mother who received 3 embryos and 1 who received X embryos.
Discussion :
By using a reliable genetic method to identify CFTC, this study shows that CFTC can be consistently found in blood of pregnant women starting from the 5th WG and that their frequency (mean number of 2,5 CFTC per ml of blood) do not substantially change during the 1st trimester of pregnancy. Furthermore, since identification of CFTC was obtained by CFTC DNA fingerprinting in women undergoing IVF after transfer of 2 or 3 embryos, this study also discovered that the majority of these women display CFTC belonging not only to the successful pregnancy but also to the abortive pregnancy. This method is able to detect a "vanishing twin" phenomenon earlier than ultrasounds imaging of the embryonic heart.
The issue of the time of appearance of circulating fetal cells in maternal blood is an old dilemma. According to the literature, circulating fetal nucleated erythroid cells and trophoblastic cells do not persist in the maternal blood after pregnancy termination. These two types of circulating fetal cells are thus considered the ideal target to develop a non invasive method of prenatal diagnosis and study the term of their appearance in pregnant women. However, the studies in this field are hampered by two major problems: 1) these cells are very rare 2) they cannot be specifically identified by immunological labelling. As a matter of fact, fetal nucleated erythroid cells represent, in the blood of pregnant women, only 30% of the overall erythroid cells, making their specific isolation and identification very difficult and unreliable.
Of considerable recent interest are data on NI-PND aneuploidy detection using cell free fetal DNA in maternal blood. Lo et al 8,15 found that a variable amount (3.4-6.2%) of free DNA in maternal plasma was of fetal origin, and thus potentially suitable for NI-PND. Circulating free fetal DNA (ffDNA) is already used clinically for prenatal diagnosis of fetal gender and Rh(D) status. However, until recently, difficulty in verifying free fetal DNA (ffDNA) in pregnant women carrying a female fetus generated difficulty, hampering exclusion of a false negative diagnosis. Detecting fetal aneuploidy using a mixed maternal and free fetal DNA sample at present requires considerable technological effort, namely several steps of complexity and high costs. Using ffDNA alone, simultaneous diagnosis of aneuploidies and single gene disorders 16 also seems unfeasible at present. Yet this could be achieved readily using pure fetal DNA from circulating trophoblasts. While studies on circulating trophoblastic cells have lately received less attention than studies targeting ffDNA, intact fetal cells thus remain valuable, if not preferable, for NIPND. The free fetal DNA strategy is, however, expected to be easier, cheaper and more rapid for detection of fetal genetic targets which are absent in the maternal DNA, such as male fetal gender (Y-specific) and RHD positive genetic target in RHD negative mothers.
Trophoblastic cells circulation has been reported to start at the 6th WG 17 more than 20 years ago, based on immune-mediated identification. However, the same authors 18 and others 19 have then reported that the antibodies used to recognize trophoblastic cells (H315, HLA-G, placenta growth factor and neuroD2) give no specific results.
By using this method, we were able to reliably establish that CFTC are systematically found in the maternal blood starting from the 5th WG. We also found CFTC at the 4th WG in 4 out of 8 tested mothers and noticed that they are very rare at this term of pregnancy.
Our data do not show major differences in CFTC frequency at early (5th, 6th WG) as compared to later terms of pregnancy (10th and 11th WG) during the first trimester. Thus, early and reliable definitive non-invasive diagnostic of genetic disorders using CFTC in maternal blood appears as a short-term, can be reachable.
CFTC analysis by DNA fingerprinting, by using two or three informative STR markers, allows to reliably identifying fetal cells genomes, through the identification of a bi-parental contribution (one paternal allele and one maternal allele). Furthermore, this method also allows to determine which paternal and which maternal allele participates to the bi-parental contribution and allows to determine the gender of the fetus at 5th WG.
In our study of 14 pregnant women of known gestation, we demonstrated by CFTC genotyping that CFTC circulate very early during pregnancy, consistently (14/14) from the 5th gestational week onward. Our results show 2.5 CFTC per ml during the first 12 WG.
Thus, DNA fingerprinting is able to distinguish CFTC derived from different embryos. In this study, CFTC belonging to different embryos were found in mothers undergoing IVF for the first time, demonstrating that we identified CFTC related to the different embryos transferred after IVF. As a matter of fact, we regularly identified in a subtype of CFTC, the genotype of the live born child, when available. The observation of CFTC with different genotypes seems to be related to the study of mothers undergoing IVF and transfer of two or three embryos.
Our molecular data clearly indicate that CFTC are frequently spread into the maternal blood both from the "successful" and from the abortive pregnancy which follow the transfer of multiple embryos. Thus, singleton pregnancies after two or three embryos transfer are often associated with an abortive pregnancy which is not detectable by routine investigations. These miscarriages are expected to occur before the 6th WG, when the presence of two hearts is investigated by ultrasounds. Actually, among the 11 mothers of this study having CFTC with at least two different genotypes and thus a vanishing twin phenomenon, ultrasounds analysis detected only one having vanishing twin and one having twins. Thus, the vanishing twin phenomenon, which is thought to occur in 10% of live born singleton 20 and which has been detected in 6.2% of cases occurs, in fact, much more frequently.
Finally, our results, showing that the real frequency of vanishing twin is higher than that detected by ultrasounds are in agreement with the observation that the incidence of first trimester bleeding increases with the number of embryos transferred 21. This method allows earlier and precise detection of abortive pregnancy one week before (at 5th WG) the ultrasounds method (at 6th WG). We conclude that the sensitivity of detection of vanishing twin by STR genotyping is more compared to the sonography method. We noticed that the number of CFTC of living embryo is more than the number of CFTC of aborted embryo in maternal blood (Table 2). Our surprising finding has two major implications: 1) If IVF is realized by transfer of two or more embryos, it will be quite "tricky" to perform an early non invasive prenatal diagnosis (NI-PND) of the ongoing pregnancy. The method will then have to plan a confirmation of the NI-PND at later (2nd trimester) time during pregnancy. The same limitation has to be expected for methods using fetal cells isolated from transcervical samples, and several studies have been carried out to investigate the obstetric outcome of pregnancies with spontaneous reduction (vanishing twin) detected by ultrasounds after IVF 22. The same studies should now be performed taking into account this new way to identify the vanishing twin phenomenon.
Conclusion :
The technique of CFTC isolation and DNA analysis by fingerprinting is very informative and powerful. Furthermore, it can be speeded up by developing high throughput biochip analyses. With this tool, we are confident that future work will bring answers to many questions asked in the past decades about circulation, persistence, elimination and clinical impact of fetal cells circulation in maternal blood.
Acknowledgement :
We thank the staff in the INSERM (National Institute for Health and Medical Research) Laboratory of Medical Genetics at the Necker Hospital, Paris - France, for their help in realization of this work. This work was supported by grants from the INSERM (Institut National de Santé et Recherche Médicale), AFM (Association Française contre les Myopathies), Anjou Mucoviscidose, ABCF Mucoviscidose, Vaincre la Mucoviscidose, Université Paris Descartes.
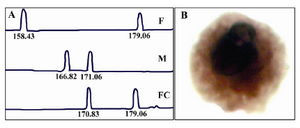
Figure 1. A) Informative STR marker applied to the paternal and maternal DNA, to the DNA of the born baby, F: father, M: mother, FC: fetal cell. B) Large KL1 positive cells identified
|
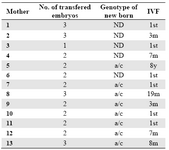
Table 1. HistoricalHistory
ND: not determined
|
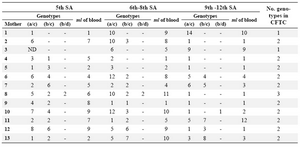
Table 2. Kinetics of appearance of the CFTC in maternal blood
|
|