Association of HLA-DRB1, DQA1 and DQB1 Alleles and Haplotypes with Common Variable Immunodeficiency in Iranian Patients
-
Amanzadeh, Amir
-
National Cell Bank of Iran, Pasteur Institute of Iran, Tehran, Iran
-
Amirzargar, Ali Akbar
-
National Cell Bank of Iran, Pasteur Institute of Iran, Tehran, Iran
-
Department of Immunology, Faculty of Medicine, Tehran University of Medical Sciences, Tehran, Iran
-
Mohseni, Nilufar
-
National Cell Bank of Iran, Pasteur Institute of Iran, Tehran, Iran
-
Arjang, Zohreh
-
National Cell Bank of Iran, Pasteur Institute of Iran, Tehran, Iran
-
Aghamohammadi, Asghar
-
Research Center for Immunodeficiencies, Pediatrics Center of Excellence, Children's Medical Center , Tehran, Iran
-
 Shokrgozar, Mohammad Ali
National Cell Bank of Iran, Pasteur Institute of Iran, Tehran, Iran, Tel: +98 21 66492595; Email: mashokrgozar@pasteur.ac.ir
Shokrgozar, Mohammad Ali
National Cell Bank of Iran, Pasteur Institute of Iran, Tehran, Iran, Tel: +98 21 66492595; Email: mashokrgozar@pasteur.ac.ir
-
National Cell Bank of Iran, Pasteur Institute of Iran, Tehran, Iran
-
Shokri, Fazel
-
National Cell Bank of Iran, Pasteur Institute of Iran, Tehran, Iran
-
Department of Immunology, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran
Abstract: Common Variable Immunodeficiency (CVID) is an antibody deficiency syndrome that often co-occurs in families with selective IgA deficiency (IgAD). This study was designed to investigate the frequency of DR and DQ loci of HLA class II region in common variable immunodeficiency (CVID) patients. Fifteen Iranian patients with CVID or IgAD (mean age 14.6±5.4, range 4-25 years; 9 male and 6 female) and 63 healthy controls were studied. Establishment of B-lymphoblastoid cell lines was performed using Epstein-Barr-virus (EBV) immortalization technique and HLA alleles were typed using polymerase chain reaction based on sequence specific primers (PCR-SSP). DRB1 alleles including DRB1 *04 (p=0.03) and DRB1 *11 (p=0.01) significantly showed higher frequency in the studied subjects. In contrast, DRB1 *301 (p=0.04) and DRB1 *07 (p=0.02) alleles were negatively associated with CVID. For DQB1 and DQA1 loci, DQB1 *0302 (p=0.047) and DQA1 *03011 (p=0.001) demonstrated high frequency in cases, while DQB1 *0201 (p=0.02) and DQA1 *0201 (p=0.01) were detected to be low when compared to controls. Haplotype analysis indicated that frequency of DRB1*04-DQB1*03011-DQA1 *03011 (p=0.02), DRB1 *11-DQB1 *03011-DQA1 *0505 (p=0.047), DRB1 *11-DQA1 *0505 (p=0.04) and DRB1*04-DQA1*03011 (p=0.02) haplotypes were significantly higher in patient group, while only the frequency of the DRB1 *07-DQA1 *0201 haplotype gene was statistically lower in control group (p=0.02). According to the results, it could be deduced that the HLA-DR and DQ loci may contribute to the pathogenesis of CVID or they might be considered as suitable markers for the possibility of the occurrence of this genetic defect.
Introduction :
Common Variable Immunodeficiency (CVID) is a heterogeneous condition characterized by B cell differentiation arrest and impaired antibody responses. The etiology of CVID has been undefined for more than 85% of CVID patients yet (1,2). CVID has been reported to affect males and females equally and it has been diagnosed in various races. Although the usual age of appearance of the disease is between the second or third decades of life, other reports described the onset of clinical complications as early as the first decade of life. On the other hand, there is a noticeable variability in the incidence of the disease in different ethnic populations and the estimated incidence of CVID in Europe, North America and other continent ranges from 1:10,000 to 1:200,000. Also, the lowest frequency of incidence has been found in Japanese (1/18000), Chinese (1/40000) and African (twenty-fold lower) descents (3-7).
In addition to the associated conditions, like recurrent infections of the respiratory and gastrointestinal systems, allergic disorders, and autoimmune symptoms, it has been found that patients with CVIDs have a 10-fold increased risk of gastric cancer (7). Evaluation of a suspected CVID usually consists of a complete blood cell count with differential white blood cell count and measurement of serum immunoglobulins level, specific antibody response and lymphocyte subsets (8). Intravenous immunoglobulin (IVIG) therapy is used in most antibody deficiencies including CVID, and also in the treatment of other autoimmune diseases (9,10).
Patients with CVID show hypo-gammaglobulinaemia in the presence of normal or near-normal B cell counts which has been named agammaglobulinemia with B cells. Also, reduced serum levels of either immunoglobulin G (IgG) or immunoglobulin A (IgA) have been reported (3).
International Union of Immunological Societies (IUIS) declared that in the severest form, patients with CVID have IgA serum levels below 0.1 g/l, IgG levels below 3.0 g/l, and usually IgM levels below 0.2 g/l (6). Furthermore, autoimmune disorders have been diagnosed mostly among relatives of selective IgA deficient individuals (SIgA-D). Most individuals with IgA deficiency had showed higher rate of autoimmunity (10%) in one of their first-degree relatives compared to an estimate of 5% in the general population (8).
Furthermore, there is an immature phenotype with the expression of IgM and IgD in CVID/SIgA-D, and they usually cannot fully develop into IgA-secreting plasma cells. Moreover, abnormalities in the cytokine network such as lack of IL-2, IL-4, IL-5, IL-10, IL-13, TGF-β, and alteration of IL-21 gene have also been proposed to play role in IgA deficiency (10,11).
Furthermore, Ig secretion by CVID B cells has been induced following infection with Epstein-Barr virus (EBV) (12). Immunoglobulin A deficiency (IgA-D) is another prevalent humoral immunodeficiency in Caucasians, but is often asymptomatic in CVID patients and B cells are affected. As mentioned, CVID may include deficiencies in other immunoglobulins as well, such as IgA and IgM deficiencies, although these deficiencies are more frequently associated with a group of other primary immunoglobulin deficiencies like agammaglobulinemia and Severe Combined Immunodeficiency (SCID). In contrast to the CVID patients, SCIDs show defects in both cellular and humoral parts of the immune system. Other components of the immune system may be normal in CVID and T-cells, the type of white cells responsible for cellular immunity, are usually manufactured at normal levels in the same individuals who have CVID and IgA deficient, although certain cell signaling components may be absent (13).
A hypothesis was that CVID and selective IgA deficiency syndrome may reflect a common underlying genetic defect because CVID and IgA-D both share clinical features (14,15). While the cases of concurrent CVID and selective IgA deficiency (SIgA-D) are occasional, familial occurrences of sIgAD and CVID have been observed in approximately 20% of cases suggesting that these heterogeneous diseases are not always clearly separable and they have a common pathogenesis and some patients with IgAD later develop CVID, and family members of patients with CVID may have only selective IgAD (14-18).
Moreover, other investigation reported cases of sIgAD developing into CVID with time and occasionally vice versa, supporting the concept that IgA deficiency and CVID lie in the spectrum of the same disease, which indicates that these conditions are closely linked and can be progressive or reversible. They may represent two phenotypic variants in a spectrum associated with the same molecular defect(s) (13-17). The etiology of CVID and IgA-D is unknown but different prevalence in various ethnic groups and familial clustering of the disorder (19,20) suggest involvement of unidentified susceptibility gene(s) in arresting B cell differentiation pathways (21,22), impairing T cell-mediated cell signaling and/or isotype class switching (23,24).
Associations between IgA deficiency and certain Major Histocompatibility Complex (MHC) alleles and haplotypes have been suggested. Furthermore, studies of families with multiple cases of sIgAD and CVID have revealed that susceptibility to CVID or IgA deficiency may be correlated with specific alleles of HLA class II genes locating in the MHC region (25-27).
The aim of the present study was to investigate whether susceptibility to CVID is associated with HLA class II alleles or haplotypes in Iranian population.
Materials and Methods :
Subjects: Heparinized peripheral blood was collected from 15 Iranian CVID patients consisting of 6 females and 9 males ranging from 4 to 25 years old (the mean age was 14.6±5.4 years) and 63 age matched healthy controls with no related disorder.
The diagnosis of CVID was based on reduction or lack of major serum immunoglobulin classes (panhypogamma-globulinemia) in serum, recurrent bacterial infections which involved different organs (ears, eyes, sinuses, nose, bronchi, lungs, skin, gastrointestinal tract, joints, bones, CNS and parotid glands), enlarged lymph nodes and loss of proteins from kidneys. All subjects and their families gave us their informed consent before their inclusion in this study.
EBV-immortalization of human B-cells: Establishment of B-lymphoblastoid cell lines was performed using EBV immortalization technique as described previously (28).
Briefly, peripheral blood mononuclear cells (PBMCs) were separated from heparinized peripheral blood by Histopaque (Sigma-USA) density gradient centrifugation and then transformed with EBV which had been produced by the B95.8 marmoset cell line (NCBI-C110; National Cell Bank of Iran, Pasteur Institute of Iran).
In this regard, the Peripheral Blood Mononuclear Cells (PBMCs) were re-suspended in the filtered supernatant of EBV containing Marmoset B95.8 cells. After 90 min incubation at 37ºC under 5% carbon dioxide with periodic agitation, the cells were washed with RPMI-1640 medium (Gibco BRL, Scotland) and again re-suspended in the same medium supplemented with 10% heat-inactivated fetal calf serum (FCS) (Gibco, Scotland) supplemented with 100 IU/ml penicillin, 100 g/ml streptomycin and 2.5 µg/ml Amphotericin-B (PAA, Austria).
The culture medium also contained 1 µg/ml cyclosporine A (CsA) (Sandoz, Switzerland) to prevent proliferation of memory T-cytotoxic cells. Outgrowth of immortalized B-cells was observable within 14 days post infection (25). Immortalized Lymphoblastoid B Cell Lines (LCLs) were then expanded and stored in Liquid Nitrogen Tank (LNT) after microbial quality control tests.
DNA extraction and HLA typing: First, EBV transformed LCLs were grown in RPMI-1640 containing 10% Fetal Calf Serum (FCS) at 37C and 5% CO2 atmosphere followed by genomic DNA extraction.
Briefly, genomic DNA was isolated from proteinase-K-treated lymphoblastoid cell lines by salting out method and HLA typing was performed by PCR amplification with low resolution sequence-specific primers (PCR-SSP) according to Olerup and Zetterquist procedure (29).
The HLA alleles were determined by molecular DNA typing of the MHC class-II HLA alleles in the DRB1, DQB1 and DQA1 loci in groups of CVID patients and control subjects. The PCR products were analysed by agarose gel electrophoresis investigating the absence or presence of amplified DNA fragments were visualized after staining with the ethidium bromide by agarose gel electrophoresis. The alleles were typed according to linkage among Iranian DRB1, DQA1, and DQB1 alleles.
Statistical analysis: Frequency values were compared for statistical significant by 2 X 2 contingency tables, using Chi-square analysis after Fisher exact test (one-tailed), as appropriate. The odds ratio (OR) with 95% confidence intervals (CI) was calculated and a value of p<0.05 was defined as statistically significant. Descriptive data are displayed as enumerations, percentages, or mean±1 SD.
Results :
Alleles frequency: Results of the HLA DRB1, DQB1 and DQA1 alleles typing in CVID patients and control group have been summarized in tables 1, 2 and 3, respectively. The data indicate that the gene frequency was higher about DRB1 alleles, because including DRB1 *04 (p=0.03) and DRB1 *11 (p=0.01) found to be significant.
On the contrary, DRB1 *301 (p=0.04) and DRB1 *07 (p=0.02) alleles were significantly decreased in CVID cases comparing to controls. For DQB1 and DQA1 loci, an increase were observed for the two alleles, the DQB1 *0302 (p=0.047) and DQA1 *03011 (p=0.001) showed an increased genotype at high frequencies. Two other alleles, DQB1 *0201 (p=0.02) and DQA1 *0201 (p=0.01) were detected at low frequencies in patients group. Most of the differences were significantly associated with CVID pathogenesis.
Haplotype frequency: HLA haplotype frequencies are displayed in tables 4 and 5. Haplotype analysis showed that the frequencies of haplotypes DRB1 *04-DQB1 *03011-DQA1 *03011 (p=0.02), DRB1 *11-DQB1 *03011-DQA1 *0505 (p=0.047), DRB1 *11-DQA1 *0505 (p=0.04) and DRB1 *04-DQA1 *03011 (p=0.02) were significantly higher, while only the frequency of the DRB1 *07-DQA1 *0201 haplotype gene was statistically lower in comparison to normal population (p=0.02).
Discussion :
Common variable immunodeficiency and IgA deficiency are heritable disorders that have been observed in immediate relatives and familial inheritance of either SIgAD or CVID occurs nearly in about 20% of cases (30). The MHC region located on chromosome 6 contains important polymorphic loci encoding different antigens that play a role in failure of B cell differentiation. HLA class-II molecules, which are expressed on the surface of APCs, present processed peptide fragments to T-lymphocyte receptors (TCR) and thereby restrict T-cell and B-cell responses to specific antigens (23,24). Several studies demonstrated that certain HLA alleles or haplotypes may confer susceptibility to CVID (15,26,31). However, some could not find any significant association for this locus (20).
In the present study, association of CVID with DRB1, DQB1 and DQA1 loci of the HLA region was investigated by DNA molecular typing, which allows the identification of previously serologically undefined specificities.
There were positive and negative significant associations for DQ and DR loci in HLA class II region specifically for DRB1 *04 (p=0.03), DRB1 *11(p=0.01), DQB1 *0302 (p=0.047), DQA1 *03011 (p=0.001) alleles. It was also observed that HLA-DRB1 *301 (p=0.04), DRB1 *07(p=0.02), DQB1 *0201 (p=0.02) and DQA1 *0201 (p=0.01) alleles occurred with decreased frequency among patients compared to control group, suggesting induction of resistance to the disease in the normal population.
The observed frequencies for all HLA antigens and alleles in control group were in accordance with those of a typical Caucasian population (32).
Unlike some studies that indicated a high frequency for DQB1 *0201 in patients (Table 6) (15,25,33,34), we observed a negative association for this allele in our study. It is interesting to note that the two DR7 (DRB1 *0701) and DQ2 (DQB1 *0201) alleles with negative associations in our last CVID study ((p<0.001 & p<0.05, respectively) were also in accordance with the data represented in this study (both with p<0.02) (32). For MHC haplotypes carrying HLA-DRB1 *07, Emilio G et al suggested an association in Spanish patients (35).
While based on our findings, “DRB1 *07-DQA1 *0201” haplotype showed a low frequency in patients. Moreover, another study reported more than 20% of individuals carried the HLA DR7- DQ2 haplotype, also they have observed a positive association for DR3, DR7 and DQ2 alleles which is in contrast with our results (34-37).
To evaluate the MHC susceptibility locus, most studies have focused on the B8-DR3 haplotype due to its prevalence in Caucasian CVID and IgAD patients. However, in Spanish patients HLA-DR1 and HLA-DR7 were shown to have higher frequencies than HLA-DR3 (25), which reflects the distribution of HLA-DR alleles and extended haplotypes in southern Europe (38). Furthermore, DRB1 *0102 allele and the B14-DR1 extended haplotype were more common among Italian CVID patients (31,39). Another study, with similar findings to ours, suggested that lower frequency of DRB1* 1501 conferred protection in this disorder (40).
In the present study, DRB1 *04-DQB1 *03011-DQA1 *03011 (p=0.02), DRB1 *11-DQB1 *03011-DQA1 *0505 (p=0.047), DRB1 *11-DQA1 *0505 (p=0.04) and DRB1 *04-DQA1 *03011 (p=0.02) haplotypes were observed in patients sharing susceptibility to CVID individuals.
However, DRB1 *07-DQA1 *0201 (p=0.02) haplotype was less frequent versus non-affected individuals. Among the 15 patients, only one (6.6%) had DRB1 *07-DQA1 *0201 haplotype, whereas 22 of the 63 healthy controls (34.9%) showed this haplotypes (p-value=0.02, OR: 0.13).
Several HLA-DR and DQ alleles and haplotypes have been identified to be associated with CVID/IgA deficient individuals in different racial populations (Table 6) (15,19,31,34-37,40-45).
In general the complexity of the Linkage Disequilibrium (LD) and the density of genes make the detection of the causative variants a challenging task in the HLA region. Based on the results we believe that susceptibility and resistance to IgA-D and CVID are correlated with alleles of the DRB1, DQB1 and DQA1 genes.
We hypothesize that the HLA class II molecules themselves are involved in the pathogenesis of IgA-D and CVID. Although the mechanism(s) of their effect is not known yet, it might act through auto-reactive T cells or by insufficient help from class II-restricted CD41 lymphocytes in T-cell-dependent B-cell responses, possibly due to inadequate response to infectious agents. Our different results from previous studies may be explained by the heterogeneous nature of these immunodeficiencies and/or environmental effects.
In addition, some researchers reported that certain MHC haplotypes with high frequencies found in immunodeficient patients were also observed in normal members of the family background. In another word these evidences suggest that the presences of these MHC haplotypes alone are not sufficient for appearance of the defects. Strong linkage disequilibrium between the different MHC loci, as was mentioned, makes it extremely difficult to determine the precise genes or haplotypes conferring susceptibility to these two diseases. The data strongly suggest the need for comprehensive HLA typing of large populations of CVID and IgAD patients, additional SNP mapping, and re-sequencing of candidate genes in MHC region.
As a final point, the method which we used in the present study to determine HLA alleles is very notable. B-cell immortalization with EBV technique leads to growth and purity of B lymphocytes. The purity of the B-cells resulted to significantly increased frequency of class-II antigens, which subsequently improves the accuracy of the classical HLA class-II detection assay (46).
Conclusion :
Considering previous data and the results of present study, differences in HLA association of IgAD and CVID compared to the other populations strongly implicate a genetic background of the disease. Furthermore, different alleles or haplotypes within the HLA class II region might be involved in the pathogenesis of these disorders and they may appear as markers for the possible genetic defect in different ethnic groups. This indicates the importance of studies to identify the associated HLA antigens in a large number of ethnic populations. However, further studies are needed.
Acknowledgement :
We are most grateful to Dr. Ali Jahanian and Ms. Zahra Safari for their help. This study was financially supported in part by a grant from Pasteur Institute of Iran.
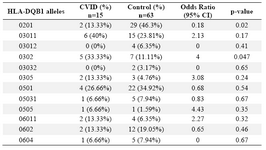
Table 1. HLA-DQB1 allele’s frequency
|
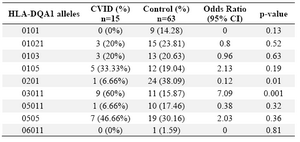
Table 2. HLA-DQA1 allele’s frequency
|
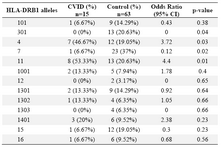
Table 3. HLA-DRB1 allele’s frequency
|
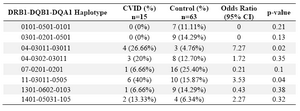
Table 4. HLA- DRB-DQB-DQA haplotype’s frequency
|
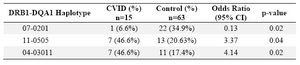
Table 5. HLA- DRB1-DQA1 haplotype’s frequency
|
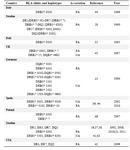
Table 6. HLA alleles or haplotypes expressed in CVID/ IgA deficiency patients from different countries
|
|