Analysis of SLC26A4 Gene in Individuals with Non Syndromic Hearing Impairment in Relation with GJB2 Associated Mutations
-
Rajalakshmi , Krishna
-
Department of Audiology, All India Institute of Speech and Hearing, Naimisham Campus, Manasagangothri, Mysore, India
-
School of Rehabilitation and Behavioral Sciences, VMRF (DU) Aarupadai Veedu Medical College Pondicherry, , India, India
-
Thirunavukkarasu, Jayakumar
-
Department of Speech-Language Sciences, All India Institute of Speech and Hearing, Naimisham Campus, Manasagangothri, Mysore, India
-
Ambika Vikraman , Meenu
-
Department of Audiology, All India Institute of Speech and Hearing, Naimisham Campus, Manasagangothri, Mysore, India
-
Department of Audiology Taluk Head Quarters Hospital, Kottarakara India , Kerala, India
-
Maruthy , Santosh
-
Department of Speech-Language Sciences, All India Institute of Speech and Hearing, Naimisham Campus, Manasagangothri, Mysore, India
-
Sylvester , Charles
-
Unit for Human Genetics, All India Institute of Speech and Hearing, Naimisham Campus, Manasagangothri, Mysore, India
-
 Kundapur , Rajesh
Unit for Human Genetics, All India Institute of Speech and Hearing, Naimisham Campus, Manasagangothri, Mysore, India 570006, Tel: +91 9156134466; E-mail: rajeshkundapur123@gmail.com
Kundapur , Rajesh
Unit for Human Genetics, All India Institute of Speech and Hearing, Naimisham Campus, Manasagangothri, Mysore, India 570006, Tel: +91 9156134466; E-mail: rajeshkundapur123@gmail.com
Abstract: Background: Hearing Loss (HL) is the most common sensory disorder. HL commonly ranges from mild to severe. Persons with HL face difficulty in hearing conversations or sounds through one ear or both ears, which impacts one’s ability to interact with others. Hence it is a communicable disorder that makes people socially isolated, lonely, and frustrated. HL in children severely affects language development. The people who are referred to as 'Deaf' with very little or no hearing capabilities, are considered as having profound hearing loss. More than 124 genes are causative for Non-Syndromic HL (NSHL) with varying inheritance, among which the SLC26A4 mutations are the second commonest cause of hereditary HL across the globe.
Methods: Samples from 70 NSHL patients were analyzed through Next-Generation Sequencing (NGS) and generated five pathogenic variants [N246fs (rs918684449), K564fs (rs746427774), F122fs, V239D (rs111033256), T721M (rs121908363)] each with frequency of 1.42%. Three missense variants [S399P (rs747431002), L597S (rs55638457), and G6V (rs111033423)] were reported under the "uncertain" category. All the collected samples were further genotyped to look for the possibility of having GJB2 and HL-associated mutations.
Results: Out of five SLC26A4 pathogenic mutations N246fs (rs918684449) and K564fs (rs746427774) were observed in samples which were positive for GJB2-HL associated candidate mutations [W24X (rs104894396), Q124X (rs397516874) and W77X (rs80338944)]. Similarly, pathogenic variants F122fs, V239D (rs111033256) and T721M (rs121908363) were observed in patient samples which were negative for GJB2-HL associated mutations.
Conclusion: Our data will expand the list of variants underlying NSHL and encourage further genotype SLC26A4 gene concerning the south Indian population with a large sample size.
Introduction :
Among different sensory disorders, Hearing Loss (HL) is the most common one, and over 5% of the world’s population suffers from one or more forms of hearing impairment 1. On average, abnormal changes in genetic makeup are solely responsible for 50% of hearing impairment 2. HL commonly ranges from mild to severe. People with HL face difficulty in hearing conversations or sounds through one ear or both ears, which impacts one’s ability to interact with others. Hence it is a communicable disorder that makes people socially isolated, lonely, and frustrated. HL in children severely affects language development. People who are referred to as 'Deaf' with very little or no hearing capabilities, are considered as having profound hearing loss. More than 124 genes are found to be causative for Non-Syndromic Hearing Loss (NSHL), with varying inheritance such as 51 genes for DFNA or autosomal dominant, 78 genes for DFNB or autosomal recessive, and 5 genes for DFNX or X-linked inheritance 3. In addition, some mutations within mitochondrial DNA are associated with HL 4. Interestingly, mutations in these genes do not occur at the same frequency across different populations or ethnic groups.
The SLC26A4 gene or Solute carrier family 26, member 4 encodes pendrin, a highly inner ear localized anion transporter 5. SLC26A4 mutations are the second most common cause of hereditary hearing loss across the globe 6. SLC26A4 mutations include non-syndromic recessive deafness and Pendred syndrome which is a combination of sensorineural hearing loss and goiter. LitVar listed 289 SLC26A4 variants including 50 with HL association 7. SLC26A4 genotyping has become an important part of molecular genetic testing for HL. In spite of numerous studies, the association of SLC26A4 with HL in different populations remains elusive. This is due to the heterogeneity of the studied cohorts and/or methods of analysis, and studies that either target screening of only the most common SLC26A4 mutations or coding and adjoined regions or the entire SLC26A4 gene 8.
Studies have shown variations in frequency distribution of SLC26A4 linked to HL among different populations across India. Five SLC26A4 mutations (G6V, I363V, S399P, I455F, and K715) were reported from the West Bengal population, and variant Y556X was detected in the Jammu and Kashmir geographic location 9. Variants S90L, V239D, V359E, G389fs, T410M, N457K, and p.K715N were reported from South India 10. The variant K715N was reported in both Jammu and Kashmir 9 and in the South Indian population 10. However, given its high heterogeneity, the number of new impairment causing gene variants for NSHL continues to grow.
The recent technological advances in target enrichment methods and Next Generation Sequencing (NGS) have overcome the barriers possessed by the earlier methods such as Sanger sequencing and laid the path for comprehensive analysis of all the known genes causing NSHL. Recently, Lin et al demonstrated the use of NGS in genetic testing of hearing impaired patients with non-confirmative GJB2 genotypes on conventional genetic examinations 11. In view of this, we have performed whole SLC26A4 gene sequencing by NGS among the South Indian patients with NSHL. Further, we investigated GJB2-associated mutations in individuals who are positive for SLC26A4 pathogenic mutations.
Materials and Methods :
Subjects, sample collection and NGS analysis: This study was carried out at the Unit for Human Genetics at the All India Institute of Speech and Hearing. Institutional ethical clearance was obtained prior to commencing the study (Ethical clearance number SH/ CDN/ARF-Aud-2/2017-18). Seventy unrelated individuals (n=40 males, n=30 females) visiting our institute for hearing impairment were enrolled for the study. Parents of NSHL children were interviewed regarding the pre, peri and postnatal history, family history and other related medical records and explained the purpose of the study. Subjects with bilateral, severe to profound hearing loss with no birth trauma, neonatal infections, physical damage or any other known etiology that can cause HL were included in the study.
After receiving the informed consent, an approved and structured questionnaire was used to obtain the general demographic information such as age, severity, type with the education and rehabilitation history. To determine the degree of HL, audio logical tests including Behavioral Observation Audiometry (BOA), Pure Tone Audiometery (PTA), Speech Audiometry (SA), Tympanometry, Acoustic Reflex Threshold (ART), Distortion Product Otoacoustic Emissions (DPOAEs) and Transitory Evoked Otoacoustic Emission (TEOAEs) were performed. All audio logical assessment tests were performed in sound treated rooms as per the standards of ANSI S3.1 (1999).
Their blood samples were collected between January 2018 to March 2019 and screened for variants in the SLC26A4 gene through NGS technology. The current study employed MiSeq, Illumina's integrated NGS instrument for data acquisition. The *.bcl files generated through the machine were uploaded to BaseSpace platform (https://basespace.illumina.com) which converts it into FastQ files. It is with the help of germline Dragon pipeline, a part of BaseSpace, suite that converts it into VCF files. Pathogenic and other significant variants reported in the VCF files under ClinVar column were selected further (Figure 1).
Results :
SLC26A4 genotyping: Variant analysis of SLC26A4 gene among study population was carried out. The Miseq Illumina platform generated 254 variants which included 15 missense variants, 5 synonymous variants, 9 frame shift mutations, 2 in-frame insertions and 222 variants pertaining to intronic and other regions (Supplementary data Table S1). Most significant variants with dbSNP numbers and ClinVar accession numbers were listed in table 1. Interestingly, variants N246fs (rs918684449), K564fs (rs746427774), F122fs, V239D (rs111033256), and T721M (rs121908363) were the pathogenic mutations observed in the present study each with frequency of 1.42%. Three missense variants [S399P (rs747431002), L597S (rs55638457) and G6V (rs111033423)] were reported under "uncertain" category. One benign variant I455F (rs375576481) was observed with frequency 4.28%. Three intronic variants (rs368970459, rs55701254 and rs753586849) with frequency of 1.42%, and one 3’ UTR variant rs2712218 with frequency of 81.42% was also evident by the current study.
Association of GJB2 variants: All the collected samples were further genotyped to look for the possibility of having GJB2 and HL associated mutations. Out of five SLC26A4 pathogenic mutations, N246fs (rs918684449) and K564fs (rs746427774) were observed in samples which were positive for GJB2 candidate mutations [W24X (rs104894396), Q124X (rs397516874) and W77X (rs80338944)]. Similarly, pathogenic variants F122fs, V239D (rs111033256) and T721M (rs121908363) were observed in samples which were negative for GJB2 associated mutations [(W24X (rs104894396), Q124X (rs397516874) and W77X (rs80338944)].
Discussion :
At present, there are more than 124 genes related to deafness, many of which are involved in inner ear function, with mutations affecting the physiology and structure of the inner ear. The most commonly identified genes are GJB2, GJB6, SLC26A4, OTOF, MYO15A, MYO7A, POU3F4, TMC1, CDH23 and mitochondrial 12S rRNA 12. SLC26A4 mutations are the second commonest cause of hereditary HL across the globe 6. It is a major gene identified to be causative for deafness among Chinese population 13. SLC26A4 gene found to be located at 7q31, and contains 21 exons and encodes ‘pendrin’ with a 780 amino acid residues acting as a polytopic transmembrane protein 5. Pendrin is essential for endolymphatic fluid resorption in the inner ear 14. Mutations in SLC26A4 may lead to an imbalance of ion circulation in internal lymphatic fluid, resulting in deafness. The variations in GJB2 and SLC26A4 in infants with HL among Chinese population with positive genotypes, account for 77.6% (52 of 67) of cases 15. As per the Indian scenario, SLC26A4 associated variants were also reported from West Bengal, Jammu and Kashmir 9 and South Indian region 10.
Despite numerous studies, the association between SLC26A4 and HL remains elusive in different populations due to heterogeneity of cohorts and/or methods of analysis. The screening method either targets only the most prevalent mutations of SLC26A4 or coding and adjacent regions 8. Our variant analysis of the SLC26A4 gene using NGS revealed five pathogenic variants [N246fs (rs918684449), K564fs (rs746427774), F122fs, V239D (rs111033256), and T721M (rs121908363). Among the five variants V239D mutation was previously reported in South Indian cohort 10.
Due to the high prevalence of GJB2 and SLC26A4 mutations and the rich literature on this topic, variants in these two genes are studied. To establish an association between SLC26A4 and GJB2, all the collected samples were further genotyped to look for the possibility of having GJB2 and HL-associated mutations. Out of five SLC26A4 pathogenic mutations, N246fs (rs918684449) and K564fs (rs746427774) were observed in samples that are positive for GJB2 candidate mutations [W24X (rs104894396), Q124X (rs397516874), and W77X (rs80338944)]. Similarly, pathogenic variants F122fs, V239D (rs111033256), and T721M (rs121908363) were observed in samples that were negative for GJB2 [(W24X (rs104894396), Q124X (rs397516874), and W77X (rs80338944)] associated mutations. A genetic counsellor or risk assessor could benefit from these findings if they work with families living in this region of India.
Conclusion :
In conclusion, we performed NGS analysis of the SLC26A4 gene among the South Indian cohort in comparison with GJB2 associated variants. Our data expands the list of variants pertaining to the SLC26A4 gene underlying NSHL and encourage further genotyping of the SLC26A4 gene with respect to the South Indian population with a large sample size.
Statement of Ethics :
All the study procedures were adhered to the principles of the Declaration of Helsinki and approved by the Institutional Ethical Committee (SH/CDN/ARF-Aud-2/2017-18).
Data availability statement: The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgement :
The authors would like to thank Dr. M Pushpavathi, Director and Dr. SR Savithri, former Director of All India Institute of Speech and Hearing for granting permission to conduct the study. We also thank Dr. N B Ramachandra and Dr. JS Jayashankar Rao for their valuable suggestions.
Conflict of Interest :
The authors have no conflicts of interest to declare.
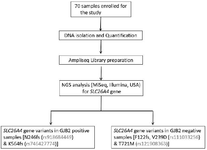
Figure 1. The overall workflow and detailing of different stages and processes used in the current investigation.
|
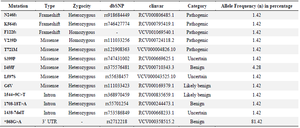
Table 1. Variants observed in SLC26A4 gene among the South Indian NSHL patients
UTR=Untranslated Region.
|
|