Producing a Mammalian GFP Expression Vector Containing Neomycin Resistance Gene
-
Izadi, Manizheh
-
Department of Medical Genetics, Faculty of Medicine, Tehran University of Medical Science , Tehran, Iran
-
Abiri, Maryam
-
Department of Medical Genetics, Faculty of Medicine, Tehran University of Medical Science , Tehran, Iran
-
 Keramatipour, Mohammad
Ph.D., Department of Medical Genetics, Faculty of Medicine, Tehran University of Medical Science, Tehran, Iran, P.O. Box: 14155-6447, Tel: +98 21 88973655, Fax: + 98 21 88953005, E-mail: keramatipour@tums.ac.ir
Keramatipour, Mohammad
Ph.D., Department of Medical Genetics, Faculty of Medicine, Tehran University of Medical Science, Tehran, Iran, P.O. Box: 14155-6447, Tel: +98 21 88973655, Fax: + 98 21 88953005, E-mail: keramatipour@tums.ac.ir
-
Department of Medical Genetics, Faculty of Medicine, Tehran University of Medical Science , Tehran, Iran
Abstract: The green fluorescent protein (GFP) was originally isolated from the Jellyfish Aequorea Victoria that fluoresces green when exposed to blue light. GFP protein is composed of 238 amino acids with the molecular mass of 26.9 kD. The GFP gene is frequently used in cellular and molecular biology as a reporter gene. To date, many bacterial, yeast , fungal, plants, fly and mammalian cells, including human, have been created which express GFP. Martin Chalfie, Osamu Shimomura, and Roger Tsien were awarded the 2008 noble prize in chemistry for their discovery and development of GFP. In many studies on mammalian cells, GFP gene is introduced into cells using vector-based systems or a recombinant virus to track the location of a target protein or to study the expression level of the gene of interest, but in these studies there is no selection marker to normalize transfection. According to the importance of neomycin gene as a selection marker in mammalian cells, we aimed to produce a GFP expression vector that contains neomycin gene. GFP gene was separated from pEGFP-N1 vector and was inserted in the back-bone of pCDNA3.1/His/LacZ vector that contained the neomycin gene. The resulted vector contained GFP beside neomycin gene.
Introduction :
In the 1960s and 1970s GFP along with the separate luminescent protein Aequorin, was purified from Aequorea Victoria and was characterized by Osamu Shimomura (1, 2). Its ability as a tool for molecular biology re-search was recognized in 1992, when Douglas Prasher cloned wtGFP and reported its nucleotide sequence (3). The first reported crystal structure of GFP was that of the S65T mutant by Remington group (4). One month later, the wild type GFP structure was des-cribed in Nature Biothec (5).
The availability of GFP and its derivatives has thoroughly redefined fluorescence micro-scopy and the way it is used in cell biology and other biological disciplines (6, 7). While most small fluorescent molecules such as FITC (Flourescein Isothiocyanate) are strongly phototoxic when used in live cells, GFP is usually much less harmful when illu-minated in living cells which widens its applications. GFP can be expressed in different structures, thus enabling morpho-logical distinction.
In such cases, GFP gene is inserted into the genome of organism in the region of DNA which codes for the target proteins, and is controlled by the same regulatory sequence. In cells where the gene is expressed and the tagged proteins are produced, GFP is produced at the same time (7, 8). Therefore, only those cells in which the tagged gene is expressed, will fluoresce when observed under fluorescent microscope. An-alysis of such time lapse movies has redefined the understanding of many biological pro-cesses such as protein folding, protein trans-port and RNA dynamics, which in the past had been studied using fixed material. An-other powerful use of GFP is to express the protein in small sets of specific cells. This allows researchers to optically detect specific types of cells in vitro or even in vivo (9).
Materials and Methods :
Vectors
pEGFP-N1 (Clontech Laboratories, Inc. GenBank Accession #U55762) vector contai-ning GFP gene, and pCDNA3.1/ His/ LacZ vector (Invitrogen life technologies) which includes neomycin resistant gene, were used in these experiments.
In all cloning experiments, the vector was cut by the appropriate restriction enzyme and run on a 1% agarose gel. Linear vector was extracted from the gel using Fermentas DNA extraction from agarose gel kit.
To eliminate the background recircularisa-tion, vectors with compatible ends were dephosphorylated. 3-5?g of vector was incu-bated with 20 units of calf intestine alkaline phosphotase (CIAP, from Amersham Phar-macia Biotech) at 37°C for 30 minutes.
Dephosphorylated vector was purified using the Fermentas PCR purification kit and its concentration measured by Spectrophoto-metry.
Inserts
The DNA fragments to be cloned were produced by restriction digestion of the source vector. Products were extracted from 1% agarose gel using QIA quick gel extrac-tion kit (Qiagen) and their concentrations were determined by Spectrophotometry.
Ligation reaction
Typically 100 ng of vector DNA and a 2 to 5-fold molar excess of insert were used with 200 units of T4 DNA ligase (New England Bio labs) in a total volume of 10 ?l at 16°C or 4°C (according to the recommen-dation of enzyme manufacturer) for 16 hours. 1 µl of the ligation mixture was then used to transform Top10F competent cells.
Bacterial transformation
1?l of DNA product from the ligation mixture was mixed with 50?l of Top 10F competent cells. The tubes were incubated on ice for 30 minutes, heat shocked at 42°C for 60-90 seconds and then returned to ice. After two minutes, 950?l Luria-Bertani (LB) me-dium was added to each transformation and the tubes were placed in an orbital incubator at 37°C for 1 hour, 250 RPM. Aliquots of 100 ?l of the culture were spread onto LB plates containing appropriate antibiotic (kanamycin 50 mg/ml or ampicillin 100?g/ml) and incubated at 37°C overnight.
Plasmid DNA preparation
4.5 ml of LB solution containing ampi-cillin (100 ?g/ml) was inoculated with a single colony and the suspension was grown at 37°C overnight. DNA was prepared from a 4 ml culture using the Accuprep Plasmid Extraction Kit (BioNeer, Korea).
In all cloning experiments the plasmids were analysed by restriction digestion and positive samples were sequenced to confirm the correct insertion of insert DNA into the vector. Sequencing was performed by Gen-fanavaran Company, Iran.
Result :
After transformation of E.coli Top10F strain cells with pEGFP-N1 vector and pCDNA3.1/His/LacZ vector, a number of obtained colonies were screened using res-triction enzyme digestion. According to the pEGFP-N1 and pCDNA3.1/His/LacZ vector maps (Figure 1), PvuII and EcoRI enzymes (Fermentas) were chosen for digestion of these vectors, respectively. Digestion of pEGFP-N1 plasmids with PvuII was expected to produce two fragments: 4.1 kb & 605 bp. And 8.7 kb band for linearized plasmid, was expected after digestion of pCDNA3.1/ His/LacZ plasmids with EcoRI (Figure 2). Double digestion of pEGFP-N1 vector by HindIII and NotI (Fermentas) resulted in two fragments (Figure 3).
The 781 bp fragment which contains GFP was purified from agarose gel. Double digestion of pCDNA3.1/His/LacZ vector by HindIII and NotI (Fermentas) also produced two fragments (Figure 3). The 5.353 bp fragment (the back-bone of the vector) containing neomycin resistant gene was purified from agarose gel. These two purified fragments were ligated and the ligation mixture was subsequently used for transformation of E.coli TOP10F cells. The resulted clones were screened by double digestion with HindIII and NotI. Those clones that produced two 5.3 kb and 781 bp bands after digestion were selected (Figure 4) and further analyzed by DNA sequencing. One of the confirmed clones was used for the sub-cloning phase.
Discussion :
In this study we produced a new vector for GFP expression that contains neomycin resistant gene. In the absence of a selection marker, neomycin gene in this case, the level of transfection using GFP expression vector would be different in various wells. By using this vector (GFP/Neo) transfection could be normalized. After 48 hours of transfection, adding G418 antibiotic can be used to select only cells with GFP expression and remove cells lacking this vector; which is synonym-ous with transfection normalization.
Acknowledgement :
The authors are very grateful to Dr. Kayhan Azadmanesh from Pasteur Insti-tute of Iran for providing plasmids. Many thanks also to Dr. Mehrdad Pedram from the Department of Medical Genetics, Tehran Uni- versity of Medical Sciences for his valuable advice during this work. This work was supported by grants from Tehran University of Medical Sciences.
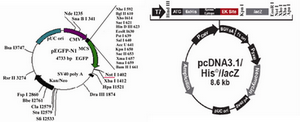
Figure 1. Vector maps of pEGFP-N1 and pcDNA3.1/His/LacZ
|
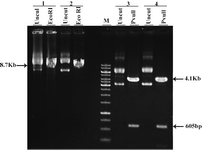
Figure 2. Restriction analysis of pEGFP-N1 and pcDNA3. 1/His/LacZ 1&2: different clones of pEGFP-N1 3&4: different clones of pcDNA3.1/His/LacZ Uncut: undigested plasmids M: 1 Kb marker
|
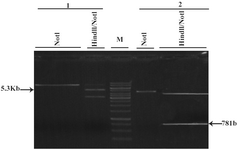
Figure 3. Extraction of GFP and Neomycin fragments after HindIII/NotI double digestion 1: pcDNA3.1/His/LacZ 2: pEGFP-N1 NotI: linearized vector with NotI 781 bp: GFP fragment of pEGFP-N1 after double digestion with HindIII/NotI 5.3 Kb: Back-bone of pcDNA3.1/His/LacZ after double digestion with HindIII/NotI M: 1 Kb marker
|
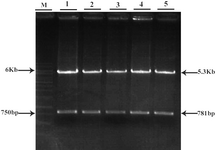
Figure 4. Restriction analysis of GFP/Neo vector
1-5: different clones of GFP/Neo vector after double digestion with HindIII/NotI
M: 1 Kb marker
|
|