Pitfalls of Restriction Enzyme Mapping Following Generation of CRISPR Constructs
-
Hassani, Mehdi
-
Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran
-
Hesami, Sara
-
Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran
-
Maroofi, Nahal
-
Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran
-
 Banan, Mehdi
Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran, Tel: +98 21 22180106; E-mail: mbbanan@yahoo.com
Banan, Mehdi
Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran, Tel: +98 21 22180106; E-mail: mbbanan@yahoo.com
Abstract: Background: The PX330 and the related PX459 plasmids are widely used for Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)/Cas9-mediated genome editing. Screening for plasmids containing the correct sgRNA template insertion is one of the most important steps in this system. Different methods for screening the sgRNA inserts have been deployed. One such method is Restriction Enzyme (RE) mapping. Restriction enzyme mapping can be used to screen for numerous plasmid recombinants simultaneously.
Methods: In this study, the sgRNA templates were initially cloned into the above PX459 plasmids. Subsequently, the accuracy of the constructs was determined by RE mapping.
Results: This method was established to screen for sgRNA-bearing PX459 plasmids. However, numerous anomalies were detected after ligation of sgRNA templates into RE digested PX459 plasmids.
Conclusion: Our data suggest that RE mapping is only appropriate as an initial screen and that the identity of all plasmids with the correctly identified RE maps should be confirmed by Sanger sequencing.
Introduction :
The Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)/Cas9 is a widely used genome editing tool 1. To perform genome editing by using this system, two components, Cas9 nuclease and a short guide RNA (sgRNA), must be present 2,3. In one platform, specific CRISPR vectors have been designed in order to express the Cas9 protein and sgRNA. The PX459 and PX330 are typical CRISPR plasmids used for this purpose. These two vectors have the same backbone, except that PX459 also contains an additional puromycin resistance (PuroR) gene 4,5.
For cloning sgRNAs templates, oligonucleotides with the appropriate Restriction Enzyme (RE) overhangs are ligated into the digested vectors. Alternatively, the entire RE site could be inserted at the ends of the oligonucleotides and then digested with the appropriate restriction enzyme(s). Next, the recombinant plasmid is created by linearizing the vector and ligating the targeted DNA fragment 5.
Next, screening for the correct ligation of inserts is necessary. There are several types of screening methods for CRISPR plasmids containing the correct sgR-NA template insertion. One such method is colony PCR. The colony PCR technique can be used to detect the presence of DNA inserts following ligation into plasmids and transformation of the ensuing ligation reactions 6. Colony PCR is a fast and cost-effective technique, but the odds of false-positive results is high. Also, there is a need to avoid false-negative results due to PCR contamination 6,7.
Alternatively, recombinant colonies can be distinguished by using restriction enzyme mapping. In this method, restriction enzymes are used to cut and determine the plasmid map based on the size of the digested fragments following gel electrophoresis. This is a reliable method that can be used to screen a large number of colonies 8. Therefore, this method is desirable to rapidly screen for likely candidate recombinant plasmids.
In this study, a restriction enzyme mapping procedure was developed in order to screen sgRNA template insertions into the PX459 plasmid. Our data suggest that this procedure can be used as an initial screen to determine the correctly inserted sgRNA templates. However, the identity of the likely candidates has to be subsequently confirmed by Sanger sequencing analysis.
Materials and Methods :
Plasmid construction: The target sequence was initially obtained from NCBI. Next, the sgRNAs were designed for upstream of the γ-globin start codon, adjacent to NGG Protospacer-Adjacent Motif (PAM) sequences (Where G is guanine and N is any nucleotide). Next, two sgRNA sequences binding just upstream of the gamma-globin start codon were designed. The design was based solely on the availability of adjacent NGG Protospacer-Adjacent Motif (PAM) sequences.
Top and bottom strands for sgRNA-1:
5’-TTTGAAGACTTCACCGTGACCCATGGCGTCT GGACTGTTTAAGTCTTCTTT
3’-AAACTTCTGAAGTGGCACTGGGTACCGCAG ACCTGACAAATTCAGAAGAAA
Top and bottom strands for sgRNA-2:
5’-TTTGAAGACTTCACCGAGCTCCTAGTCCAGA CGCCAGTTTAAGTCTTCTTT
3’-AAACTTCTGAAGTGGCTCGAGGATCAGGTC TGCGGTCAAATTCAGAAGAAA
Next, the single-stranded oligonucleotides were annealed to create double-stranded sgRNA DNA templates. For this purpose, the top and bottom strands of each oligonucleotide were denatured at 95°C for 2 to 3 min using a 100 mM NaCl annealing buffer and then annealed at room temperature (25°C) for one day.
Double stranded oligonucleotides and PX459 were digested separately with BbsI enzyme (Thermo Fisher Scientific, USA). Then, the linearized vector and oligonucleotides were gel purified using the Silica Bead DNA Gel Extraction Kit (Thermo Fisher Scientific, USA). Finally, the sgRNA templates were ligated into the PX459 plasmid (Addgene, USA) by T4 DNA ligase (Thermo Fisher Scientific, USA).
Screening: The constructs were transformed into Escherichia coli (E. coli) DH5α using the CaCl2 method 9. Plasmid extraction was performed by alkaline lysis mini-preparation method 10. The accuracy of the constructs was determined by enzyme digestion using enzymes BbsI and EcoRI followed by electrophoresis on a 1% agarose gel. Then selected plasmids potentially containing the inserted sgRNA templates were assessed by using Sanger sequencing and the U6-Forward primer, 5′-GAGGGCCTATTTCCCATGATTCC-3′.
Results and Discussion :
Two sgRNA sequences were designed based on the available PAM sequences upstream of the γ-globin start codon. The sgRNA DNA templates were then digested and cloned into the PX459 plasmid. In order to screen for the correct sgRNA template insertions via RE mapping, the plasmids were cut with the BbsI and EcoRI restriction enzymes. Digestion of plasmid PX459 with BbsI and EcoRI should create three visible bands using gel electrophoresis (Figure 1). In the recombinant PX459 harboring the sgRNA template, however, the BbsI cut site is removed and only two bands should be observed after BbsI-EcoRI double digestion (Figure 1).
Several obtained recombinant PX459 plasmids with predicted two band RE pattern were analyzed by Sanger sequencing (Figures 2A and 2B). Most of these plasmids had the correctly inserted sgRNA template. A number of these recombinant plasmids, however, were incorrectly validated based solely on their RE maps.
Some of these "incorrect recombinant plasmids" involved spurious small deletions or insertions (Table 1). In one case (sgRNA1-7), the region between the two adjacent BbsI sites was deleted and plasmid re-ligated without any sgRNA template insertion. This led to a 26 bp deletion in the plasmid. In another case (sgRNA2-22), one BbsI recognition site, which was included in the annealed sgRNA oligonucleotides, was not digested prior to plasmid insertion. This led to an unwanted 15 bp addition and the retention of a BbsI site in the construct.
Other plasmids, however, harbored large deletions and insertions spanning the BbsI recognition site--again, leading to a two band RE map pattern. In one clone (sgRNA1-1), a 4507 bp region spanning the BbsI site was deleted. In another (sgRNA2-27), a 3400 bp segment of the PX459 plasmid harboring this region was deleted. As expected, one end point of these deletions involved the BbsI recognition site. The other end, however, involved sequences 5/6 similar to that of the BbsI recognition site (5'-GAAGAC-3'). In two other clones (sgRNA1-4 and sgRNA1-15), insertions of E. coli sequences into this region were also detected.
Conclusion :
Collectively, the above results suggest that RE mapping can only be used as an initial screen for determining the correctly inserted sgRNA templates into plasmids. Screening by this method is reliable and can be used to screen a large number of samples. However, as shown, this screening may lead to the validation of incorrectly identified recombinant plasmids. Therefore, plasmids with the correct RE map should be verified via Sanger sequencing.

Figure 1. A) Schematic view of the expected PX459-sgRNA restriction enzyme map. PX459 has two cut sites for EcoRI and BbsI. B) In the PX459 containing a sgRNA template insert, the BbsI cut site is removed. C) A digestion simulation of PX459 (with sgRNA and PX459 without sgRNA) is shown using the SnapGene software (insightful Science, USA). Double digestion of PX459 with EcoRI and BbsI should result in 3 visible bands (5253 bp, 3231 bp, 669 bp) using gel electrophoresis. In PX459 plasmids with a sgRNA insert, the BbsI sites are removed and only two bands (8509 bp, 669 bp) should be observed.
|

Figure 2. A) Restriction enzyme (BbsI-EcoRI) digests several of the isolated recombinant PX459 plasmids. Some of the plasmids having a two band pattern (8509 bp and 669 bp) had the correct RE map (*), whereas others did not (#). B) Digestion simulations (BbsI-EcoRI double digestion) were performed for the plasmids highlighted in table 1 by using the SnapGene software. In sgRNA2-22, one BbsI recognition site was not digested prior to plasmid insertion. The existence of this enzyme site led to 3 bands in gel electrophoresis after BbsI-EcoRI double digestion.
|
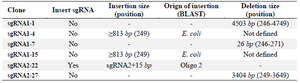
Table 1. Summary of the spurious deletions and insertions following the cloning of sgRNA templates into the PX459 plasmid
|
|