Non-Viral Transfection Methods Optimized for Gene Delivery to a Lung Cancer Cell Line
-
Salimzadeh, Loghman
-
Shiraz Institute for Cancer Research, Shiraz University of Medical Sciences, Shiraz, Iran
-
Jaberipour, Mansooreh
-
Shiraz Institute for Cancer Research, Shiraz University of Medical Sciences, Shiraz, Iran
-
Hosseini, Ahmad
-
Shiraz Institute for Cancer Research, Shiraz University of Medical Sciences, Shiraz, Iran
-
 Ghaderi, Abbas
Shiraz Institute for Cancer Research, Shiraz University of Medical Sciences, Shiraz, Iran , +98 711 230 3687; ghaderia@sums.ac.ir
Ghaderi, Abbas
Shiraz Institute for Cancer Research, Shiraz University of Medical Sciences, Shiraz, Iran , +98 711 230 3687; ghaderia@sums.ac.ir
-
Shiraz Institute for Cancer Research, Shiraz University of Medical Sciences, Shiraz, Iran
-
Department of Immunology, School of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
Abstract: Background: Mehr-80 is a newly established adherent human large cell lung cancer cell line that has not been transfected until now. This study aims to define the optimal transfection conditions and effects of some critical elements for enhancing gene delivery to this cell line by utilizing different non-viral transfection Procedures.
Methods: In the current study, calcium phosphate (CaP), DEAE-dextran, superfect, electroporation and lipofection transfection methods were used to optimize delivery of a plasmid construct that expressed Green Fluorescent Protein (GFP). Transgene expression was detected by fluorescent microscopy and flowcytometry. Toxicities of the methods were estimated by trypan blue staining. In order to evaluate the density of the transfected gene, we used a plasmid construct that expressed the Stromal cell-Derived Factor-1 (SDF-1) gene and measured its expression by real-time PCR.
Results: Mean levels of GFP-expressing cells 48 hr after transfection were 8.4% (CaP), 8.2% (DEAE-dextran), 4.9% (superfect), 34.1% (electroporation), and 40.1% (lipofection). Lipofection had the highest intense SDF-1 expression of the analyzed methods.
Conclusion: This study has shown that the lipofection and electroporation methods were more efficient at gene delivery to Mehr-80 cells. The quantity of DNA per transfection, reagent concentration, and incubation time were identified as essential factors for successful transfection in all of the studied methods.
Introduction :
Gene delivery is one of the most basic techniques of molecular biology, a technological basis for in vitro and in vivo gene therapy 1. Expression of transgenes in cell cultures provides a suitable system to determine the structure, regulation and function of a desired gene 2,3.
There are two main types of transfection strategies, viral and non-viral. Viral gene transfer methods provide the highest transfection efficacy, but have serious limitations such as the size of DNA carried in the vector, intrinsic biosafety issues, and concern for viral insertion mutagenesis 4. In comparison, although non-viral methods are less efficient, they allow for a shorter duration of transgene expression. Non-viral transfection methods are also attractive because they enable a flexible size of DNA to be transported, are less expensive, easier to prepare and generate little or no in vivo immune response 5-7.
Both chemical and physical systems are used in non-viral transfection methods. In chemical-based systems, synthetic or naturally occurring compounds such as Calcium phosphate (CaP) 8,9, DEAE-dextran 9, cationic lipids 10,11, and cationic polymers 12 facilitate the transfer of a plasmid DNA construct through the cell membrane. The efficacy of chemical non-viral gene delivery methods and their safety for cells is dependent on various factors such as the type of method, ratio of plasmid DNA to reagents, charge and size of complexes, time of exposure, type of target cell, and correct cell density 13-15. Therefore, careful optimization is required for gene delivery into the target cells in each transfection method.
Physical methods such as microinjection 16, gene gun 17,18 and electroporation 19,20 are carrier-free gene delivery techniques that employ the use of a physical force to permeate the cell membrane and facilitate intracellular transfer of naked DNA. Electroporation, as one type of physical transfection method, uses an electrical pulse to the cell to induce formation of transient pores through the cell membrane, allowing entry of the plasmid DNA into the cells 21. Factors to be considered for optimal gene transfer using electroporation are the electrical field strength and pulse duration, ionic strength of the electroporation buffer, nucleic acid concentration, cell density and viability 22,23.
It is essential to evaluate optimal conditions for gene transfer with each transfection method, as these conditions possibly differ for various cell types. In this study, we have compared the transfection efficiency of the classical methods, CaP and DEAE-dextran; two commercially available non-viral methods, superfect and lipofection; and a physical transfection method, electroporation, within the Mehr-80 cell line. Mehr-80 is an adherent human large cell lung cancer cell line with neuroendocrine differentiation that has previously been established in our institute 24. Until now no report has been published regarding gene transfection into this cell line. We have also elucidated important factors that affected the transfection efficacy using these methods.
Materials and Methods :
Cell culture: Mehr-80 cell line was cultured in RPMI-1640 medium that contained 10% Fetal Bovine Serum (FBS), penicillin (100 U/ml), and streptomycin (100 µg/ml) at 37°C in a humidified atmosphere with 5% CO2. The medium, FBS, and antibiotics were all purchased from Biosera (UK).
Plasmid preparation: To optimize and obtain highly efficient transfection, we used pEGFP-N1 (Clontech Laboratories, USA) that codes the GFP and kanamycin/ neomycin-resistant gene. GFP is commonly used as a reporter protein in gene transfection studies. For amplification, pEGFP-N1 was transformed to the E. coli-DH5α competent cells by heat shock transformation, following standard laboratory protocols. The transformed bacteria were selected on an LB-kanamycin agar plate and amplified in LB-kanamycin medium. The plasmids were purified from cultured transformed bacteria using a Plasmid Maxiprep Purification Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. Plasmid DNA was diluted in sterile water at a concentration of 1 µg/µl. For plasmid confirmation, the extracted products were cut by Hind III and Not1 enzymes and then visualized by electrophoresis in 1% agarose gel.
In addition to pEGFP-N1, we used another plasmid that encoded Stromal cell-derived Factor-1 (SDF-1:pCMV6-XL4) in a separate transfection experiment. pCMV6-XL4 was purchased from Origene Company (Rockville, MD, USA) and prepared by a method similar to pEGFP-N1, with the exception that this construct was selected by ampicillin-LB agar.
Transfection by calcium phosphate (CaP) coprecipitation: One day prior to transfection, we seeded 5×105 cells that were in the exponential growth phase into 25 cm2 cell culture flasks. Cells grew overnight to reach about 60% confluency. For CaP precipitate formation, we used a solution of 0.25 M CaCl2 (150 µl) to evaluate 2, 5, 10 and 20 µg of plasmid DNA. The CaCl2-plasmid DNA solution was gently added to an equal volume of 2X Hepes-buffered saline solution (50mM HEPES, 140 mM NaCl, 1.5 mM Na2HPO4, pH=7.05) in a 15 ml falcon tube. This mixture was maintained at room temperature for 20 min. Next, we washed the cells with serum-free RPMI-1640 medium; then, prepared precipitates were added to the washed cells and incubated for 2.5, 4, 8 or 18 hr. After defining the optimum condition for transfection, we applied a glycerol shock to determine if it could increase the transfection rate. After the last incubation, cells were exposed to 2 ml of 10% glycerol containing RPMI-1640 medium for 90 s to create the glycerol shock condition. Glycerol was subsequently neutralized by the addition of complete medium (RPMI-1640 that contained 10% serum) and the mixture was aspirated and replaced with fresh medium.
Transfection by DEAE-dextran: One day before transfection, we seeded 5×105 cells into 25 cm2 cell culture flasks. Cells grew overnight to reach about 60% confluency.
To prepare the transfection mixture, 100 µl solutions of either 10, 20, 40 or 80 µg/ml DEAE-dextran (Sigma, USA) in Tris Buffered Saline (TBS) solution were added to 0.5, 1, or 2 µg of plasmid DNA, separately, in sterile 1.5 ml propylene tubes. The mixtures were kept for 30 min at 37°C, after which their volumes were increased to 1 ml by the addition of complete medium. The mixtures were added to the cells and incubated for an additional 2 hr under cell culture conditions. We evaluated whether DMSO shock could improve DEAE-dextran transfection efficacy by the addition of 2 ml of RPMI medium that contained 10% DMSO to the cells. After incubation for 60 s, the DMSO solution was promptly removed and replaced with fresh medium.
Transfection by superfect: One day prior to transfection, 5×104 cells were seeded per well of 24-well plates in RPMI-1640 medium that contained 10% FBS. On the day of transfection, cell density reached approximately 60% confluency. To determine the optimal transfection condition, we prepared the following ratios of plasmid DNA (µg) to Superfect reagent (µl): 0.5:1, 0.5:2, 0.5:5, 1:2, 1:5, 1:10, 2:4, 2:10, and 2:20 in 100 µl RPMI-1640 medium without serum, according to the manufacturer's protocol (Qiagen, Hilden, Germany). Solutions were incubated for 10 min at room temperature. Next, 600 µl of RPMI-1640 medium that contained 10% serum was added to the mixture and cells were incubated for 1, 2 or 3 hr at standard cell culture conditions. Subsequently, transfection solution was aspirated and replaced with 1 ml complete medium per well.
Transfection by lipofection: One day before transfection, 7×104 cells were seeded per well of 24-well plates in RPMI-1640 that contained 10% FBS with no antibiotics. On the transfection day, cell confluency reached approximately 90%. For complex formation, first, 1 µg of plasmid DNA and different amounts (0.5, 1, 2, 3 or 4 µl) of Lipofectamine 2000 reagent (Invitrogen, USA) were assessed according to the manufacturer's protocol. After determining the correct amounts of Lipofectamine 2000 reagent, we evaluated either 2, 3 or 4 µg of plasmid DNA. For complex formation, plasmid DNA and Lipofectamine 2000 reagent were diluted in 50 µl of RPMI-1640 that contained no serum and antibiotics. Then, the diluted plasmid DNA and reagents were combined and incubated for an additional 25 min at room temperature. Next, we aspirated the supernatant and replaced it with 100 µl RPMI-1640 medium that contained no serum and antibiotics. Subsequently, prepared complexes were added to the cells and incubated for 6 hr under standard cell culture conditions. Finally, the transfection solution was aspirated and replaced with 1 ml complete medium per well.
Transfection by electroporation: On the day of transfection, 2×106 cells and 10 µg of plasmid DNA were diluted in 400 µl RPMI-1640 medium that contained no serum. The cell suspension was pipetted into a sterile electroporation cuvette that had a 4 mm gap. Electroporation was performed with a Gene Pulser-Xcell Electroporation Device (Bio-Rad, USA). To establish an optimal setup, in the first setting, the electroporator device was set to a voltage of 260 or 300 V and the capacitance parameter varied from 100 to 1500 µF. In the second setting, one or two pulses were applied with a 5 ms pulse length and voltage range from 600 to 960 V. After applying the electrical pulse for each situation, we added 500 µl of RPMI-1640 medium that contained 10% FBS to the electroporation cuvette, following by incubation on ice for 3 min. Finally, cell suspensions were transferred to the 25 cm2 cell culture flasks.
Detection of transgene expression and cell viability: In each method, transfected cells were incubated at 37°C with 5% CO2 for 48 hr, after which green fluorescence was detected by fluorescence microscopy (Olympus, Tokyo, Japan). Cells were detached from the flasks or plates and washed once with Phosphate Buffered Saline (PBS), and then resuspended in PBS. Transfection efficacy was quantified using flow cytometry (FACS Caliber, Becton Dickinson, USA) by scoring the percentage of cells that were successfully transfected. Since untransfected cells accounted for background fluorescence, cells only treated with each reagent and no plasmid were used as controls. Analysis of flow cytometry results was performed using FlowJo software.
Cell viability was also assessed using trypan blue staining 48 hr after each transfection method. For this purpose, adherent cells were mildly trypsinized and suspended cells in medium were mixed with 0.01% trypan blue dye, and then evaluated.
SDF-1 transfection and real-time PCR analysis: We wanted to further evaluate transfection efficiency of the above-mentioned techniques based on the intensity of the transfected gene. Thus, the optimum condition of each transfection method was selected for delivery of plasmid constructs that encoded the SDF-1 gene into Mehr-80 cells. RNA was extracted from untransfected and SDF-1 transfected cells within 48 hr after transfection by using TRIzol reagent (Invitrogen, USA). cDNA was synthesized by a RevertAid First Strand cDNA Synthesis Kit (Fermentase, Lithuanian) according to the manufacturer’s protocol. We used the Chromo 4 Detection System (Bio-Rad, USA) for real-time PCR. β-actin was the internal control. PCR reactions were performed in a total volume of 25 μl that contained 12.5 μl of 2x SYBR Green PCR Master Mix (Applied Biosystems, USA), 100 nM of each primer and 1.0 μl cDNA template in each sample. The amplification program consisted of an initial denaturation step (10 min at 95ºC) and an amplification step (15 s at 95ºC; 30 s at 37ºC; and 34 s at 60ºC for 40 cycles). The accuracy of amplification was confirmed by melting curve analysis and electrophoresis on 2% agarose gel.
The primers for the target genes were designed by Primer-Blast NCBI online software and synthesized (Bioneer, Korea). β-actin: forward (5΄-AGCACTGTGTTG GCGT ACAG- 3΄) and reverse(5΄ -GGACTTCGAGC AAGAGATGG- 3΄) and SDF-1: forward(5΄-TGC CAGAGCCAACGTCAAG- 3΄) and reverse (5΄-CAGCCGGGCTACAATCTGA A- 3΄). The relative quantification of gene expression in each sample was calculated by the 2-ΔΔCtformula.
Statistical analysis: Statistical analyses were conducted using SPSS 15.0 (SPSS Inc., Chicago, IL, USA) and GraphPad Prism 4.0 (GraphPad Software, La Jolla, CA, USA). Data were expressed as mean±SD of separate experiments in different transfection methods. The medians between pairs of groups were compared using the student's t-test. The medians between more than two groups were compared by the Kruskal-Wallis test. A two-tailed p-value less than or equal to 0.05 with 95% confidence intervals was considered statistically significant.
Results :
Transfection efficacy of the Mehr-80 cell line using the CaP method under optimum conditions after a glycerol shock, with 10 µg plasmid DNA and an incubation period of 4 hr was 8.3±1.64% (Figure 1A). The mean level of transfection without the glycerol shock under the above conditions was 6.2±0.9%. Data showed that the transfection rate was enhanced by the increase in plasmid DNA concentration from 2 µg to 5 µg during all incubation times. As shown in figure 2 in application of 20 µg plasmid DNA, the transfection rate decreased when the transfection reagent was incubated on the cells for longer time (Figure 2).
Our results showed that the highest transfection rate with the DEAE-dextran method (8.2±0.6%) was observed when we used 2 µg of plasmid DNA and 20 µg/ml DEAE-dextran in conjunction with DMSO shock (Figure 1B). Increased DEAE-dextran concentrations did not enhance transfection level in the steady state and resulted in extensive cellular toxicity as recognized by various amounts of suspended dead cells on the culture medium. Application of 80 µg/ml DEAE-dextran was not tolerable by the cells, and resulted in total cell death (Figure 2). DEAE-dextran transfection of Mehr-80 cells without DMSO shock resulted in only 3.7±1.4% efficacy under optimum conditions.
Transfection efficacy with the Superfect reagent under optimal conditions was only 4.9±0.4%, which was obtained by the application of 1 µg of DNA, 5 µl of the reagent and a 2 hr incubation period (Figure 1C). The additional increase in plasmid DNA to Superfect reagent ratio and increased incubation time was not tolerated; no increase in transfection rate was noted.
Under optimal conditions for the electroporation method, the transfection rate was observed to be 34.2±2.2%, with the application of DNA (10 µg) at a setting of 850 µF and 260 V (Figure 1E). While the efficacy rate was considerably lower with the second evaluated setting, our results suggested that 40.1±1.4% was the highest transfection efficacy obtained using the lipofection method (Figure 3). Optimal conditions for this method were determined to be the ratio of 1 µg plasmid DNA to 4 µl lipofectamine 2000 reagent, followed by 6 hr of incubation (Figure 1F). Application of additional amounts of DNA or reagent decreased transfection efficacy.
Results demonstrated that there was no significant difference in the percentage of GFP-positive cells between lipofection and electroporation (p>0.05). No significant difference was also noted between results obtained by CaP, DEAE-dextran transfection and superfect (p>0.05). Electroporation and lipofection were more efficacious than the other methods (p<0.0001).
As shown in figure 3, DEAE-dextran transfection exhibited higher efficacy than superfect (p< 0.001).
To estimate intensity of the gene expression, we compared the ratio of SDF-1 expression to β-actin under optimum conditions for the different transfection methods (Figure 4). Our results showed that unlike the percent of GFP-transfected cells, there was a significant difference in SDF-1 expression between lipofection and electroporation (p<0.0001). Levels of SDF-1 expression also differed between CaP, DEAE-dextran, and superfect (p<0.05). SDF-1 expression in electroporation technique was lower than lipofection, but higher than the other applied methods (p<0.0001; Figure 4).
When we estimated the toxicity of each transfection method with trypan blue dye at 48 hr after transfection, our results showed the following mean levels of nonviable cells: 24% (CaP), 36% (DEAE-dextran), 7% (superfect), 16% (electroporation), and 11% (lipofection).
Discussion :
The ability to transfer and suitably express an exogenous gene into mammalian cells has become increasingly important in biomedical research and therapeutic development 25-28. The transfection efficacy varies widely and is dependent upon the target cells and parameters necessary for transfection optimization that need to be promoted and specified for each method and cell type. In the present study, we determined the optimum condition of pEGFP-N1 transfection in the Mehr-80 cell line by performing transient expression assays using different non-viral transfection methods.
As expected, the different methods produced inconsistent results. In the CaP method we researched the effects of different DNA concentrations and incubation times of CaP/DNA complexes on the transfection rate of target cells. As with other studies our results have confirmed the poor transfection rate of CaP compared to newly synthesized methods, such as lipofection 7,10. According to the results, increase of DNA concentration from 2 µg to 10 µg and incubation times from 2 to 4 hr augmented transfection rate, and reached its optimum level (Figure 2). However, an inverse relationship between incubation time and transfection efficacy was observed at a high plasmid DNA concentration (Figure 2). This might be related to the increase in size of the plasmid DNA/CaP complexes and their inability to enter the cell membrane. Therefore, cell incubation time and plasmid DNA concentration were critical parameters for successful CaP transfection.
Precipitation of plasmid DNA with CaP is a common, conventional non-viral gene transfer technique. In this method plasmid DNA forms a tight complex with CaP which enters into cells by phagocytosis. This method is beneficial because of its low cost and ability to generate stable transfects in a wide variety of cell lines 29. Nevertheless, CaP transfection is completely sensitive to slight changes in buffer salt concentrations, temperature, and pH 29.
Although our results indicated no significant differences in the levels of GFP transfection between CaP, DEAE-dextran, and superfection, in comparison the DEAE-dextran transfection showed substantial cell death (Figure 3) which agreed with previous studies that other cells were used 30. Our results showed that increase of DEAE-dextran and plasmid DNA concentrations enhanced transfection efficacy. The increase in DEAE-dextran from 10 µg/ml to 20 µg/ml and plasmid DNA concentration from 0.5 µg to 2 µg improved transfection rate and achieved optimum efficacy (Figure 2). As seen with CaP transfection, there was a converse relation between plasmid DNA concentration and transfection efficacy at a concentration of 40 µg/ml DEAE-dextran (Figure 2).
The treatment of Mehr-80 cells with DMSO enhanced transfection within this method despite increased cell toxicity. DEAE-dextran is a low cost transfection reagent but because of its toxic effects and inability to create stable cell lines 9,31, it appears to be less preferable than other methods for Mehr-80 transfection.
In subsequent experiments, despite the low toxicity of the Superfect reagent, it was not possible to obtain good transfection efficacy, which was unexpected. The reason was not clear, however, it might be related to our cell line, Mehr-80. Based on the percent of GFP transfected cells, no statistically significant difference existed in terms of efficacy between superfect, CaP and DEAE-dextran, however this difference was significant when we analyzed SDF-1 expression. In comparison of flow cytometry and real-time PCR methods for evaluation of GFP and SDF-1 expression respectively, our results showed that in superfect method, despite low frequency of transfected cells, intensity or level of transgene expression per viable cell was higher than CaP and DEAE-dextran methods. These results were confirmed by brighter GFP expression in photographs of Superfect than CaP and DEAE-dextran methods (Figure 1F).
Commercially available cationic lipoplexes and polyplexes such as the Superfect reagent and lipofection are valued for their limited toxicity, ease of use and ability to transfect a wide range of cell types with relatively higher efficiencies than seen with previously mentioned methods 10,32. Based on our results and taking into consideration the price of the Superfect reagent, it seems that this method is not preferable to the other studied methods for gene delivery to the Mehr-80 cell line.
In the current study, lipofection showed the highest efficacy for transfection of Mehr-80 cells, based on the percentage of GFP-positive cells (Figure 1F) and levels of SDF-1 overexpression (Figure 3). Later evidence has suggested that using lipofection the density of transfection or numbers of transfected plasmids per cell were higher than the other methods. Cationic liposomes are known to be one of the best non-viral methods used in gene therapy. Despite the numerous benefits of cationic lipid reagents, limitations exist such as the requirements to adhere to strict conditions that include the correct cell density, sensitivity to antibiotics in the culture medium, optimization of plasmid DNA concentration, and exposure duration for every cell type 32. However, our results have demonstrated that because of high efficacy and low toxicity, lipofection is recommended as one of the best non-viral methods for gene delivery to Mehr-80 cells.
In this study, transfection rate using the electroporation method was higher than superfect, CaP and DEAE-dextran techniques. Electroporation is highly efficient for both transient and stable transfection of multiple cell types 19. However, as with chemically mediated transfection techniques, electroporation conditions are cell line dependent and should be determined experimentally. With the exception of the cost for an electroporator machine, the electroporation method is advantageous because of large scale transfection and no need for a transfection reagent. However, due to the different variable factors in transfection and time consuming optimization it might be the preferable method after lipofection. Our results have indicated that for producing higher gene delivery rates, further optimization of the program setting and application of another electroporation buffer may be necessary in future studies.
Conclusion :
In conclusion, this study showed that lipofection and electroporation were more practical and reproducible for the evaluation of gene overexpression in Mehr-80 cells. For all studied methods, we determined that the quantity of plasmid DNA per transfection, reagent concentration, and incubation time of the cells with the transfection medium were essential factors for successful transfection. Our results provided useful information for optimizing transfection in this cell line.
Acknowledgement :
This work was financially supported by a grant from Shiraz University of Medical Sciences (grant number: 87-4052) and also by Shiraz Institute for Cancer Research (grant number: ICR-100-505).

Figure 1. Photographs and flow cytometry results of transfection using different methods. Cells were analyzed for GFP expression by fluorescence microscope (20x) and flow cytometry 48 hr from transient transfection in comparison with untransfected cells. A) Untransfected Mehr-80 cells: B) CaP; C) DEAE-dextran; D) Superfect; E) Electroporation; F) Lipofectamine 2000. Data obtained from flow cytometry were analyzed using FlowJo software. Plots represent percent of positive GFP expressing cells. Transfected cell in the photographs of Lipofection and electroporations are brighter than the other techniques, which concordant with the flow cytometry results, indicate the higher density of transfectin
|
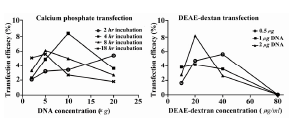
Figure 2. Different situations for optimizing CaP and DEAE-dextran method. Left figure shows effects of incubation time and DNA concentration on transfection rate in CaP method. Right figure shows effects of DEAE-dextran and DNA concentration on transfection rate of DEAE-dextran method (B)
|
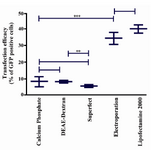
Figure 3. Comparison of pEGFP-N1 transfection efficacy in Mehr-80 cells using different methods. Data represent the mean (±SD) of flow cytometry analysis from three independent experiments in optimum situation of different transfection methods (*: p-value <0.05, **: p-value <0.01, ***: p-value <0.0001)
|
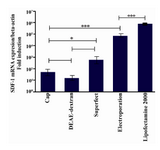
Figure 4. Comparison fold overexpression of SDF-1 after transfection of Mehr-80 cells using plasmid construct encoding SDF-1. Data represent the average expression of SDF-1 gene relative to β-actin±standard deviation of triplicate transfection experiments in optimum situation of different transfection methods
|
|